

Hyperactivation of HUSH complex function by Charcot-Marie-Tooth disease mutation in MORC2
- PMID: 28581500
- PMCID: PMC5493197
- DOI: 10.1038/ng.3878
Hyperactivation of HUSH complex function by Charcot-Marie-Tooth disease mutation in MORC2
Abstract
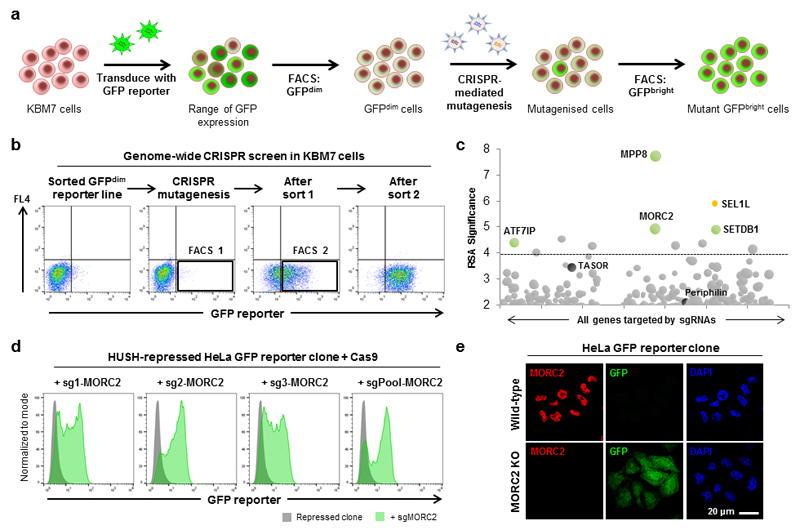
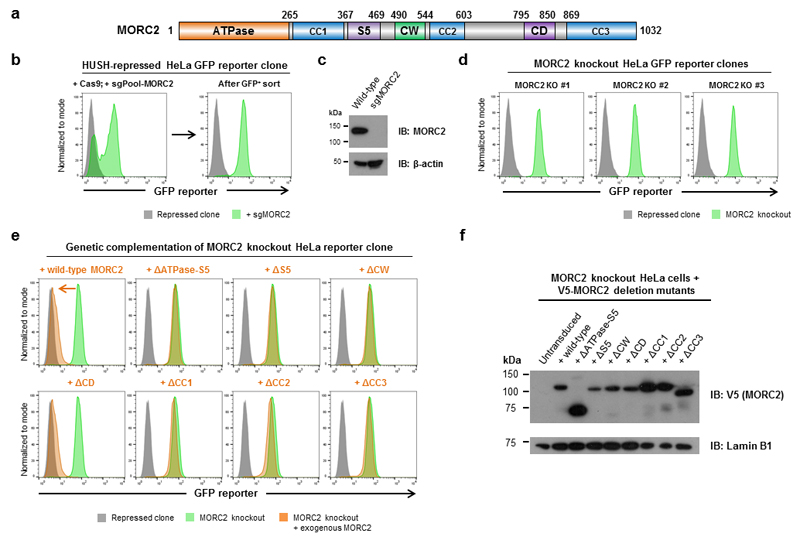
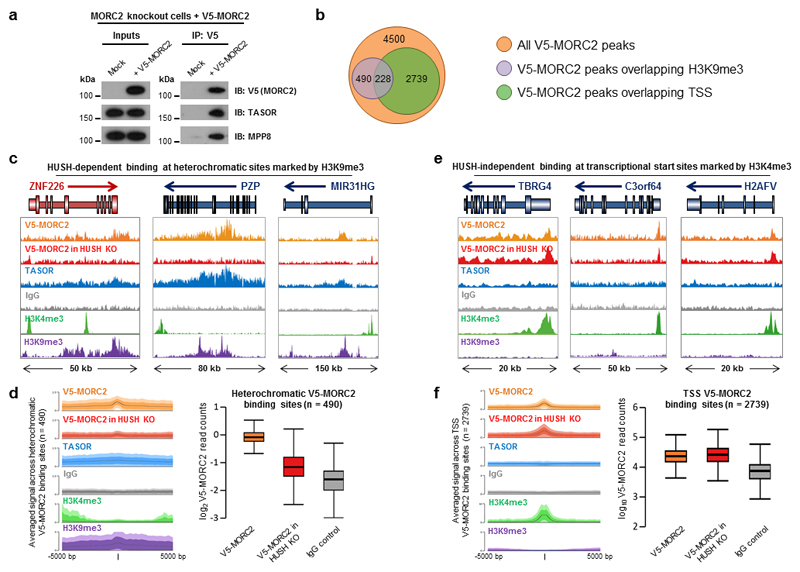
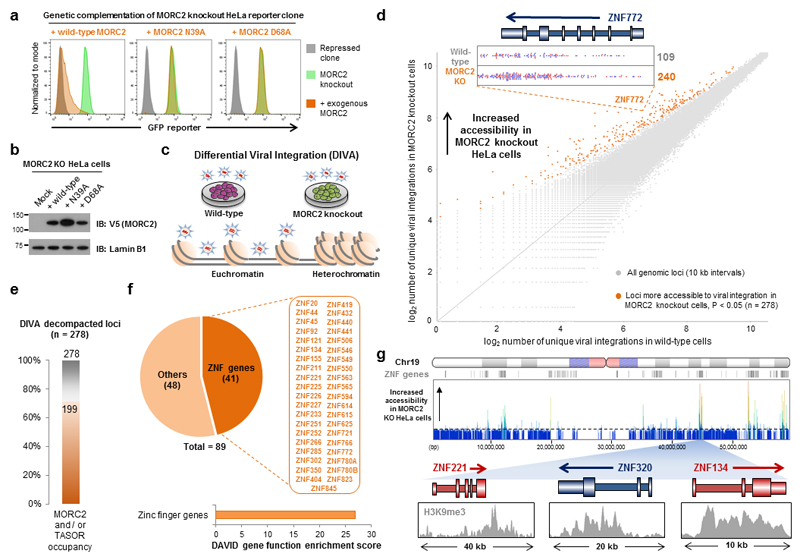
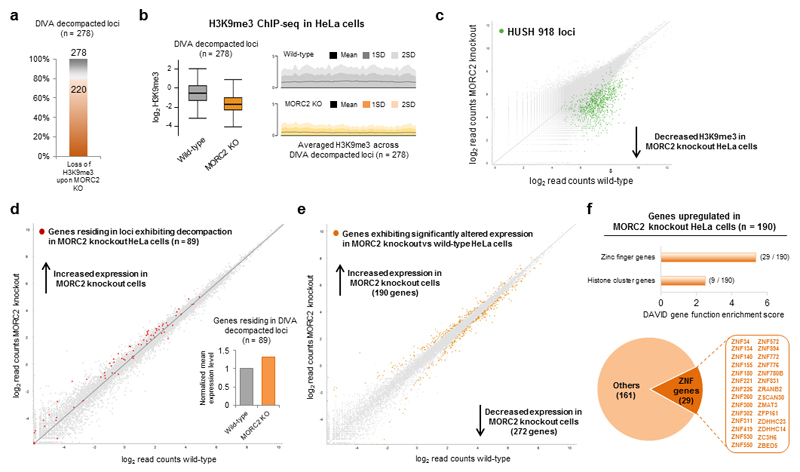
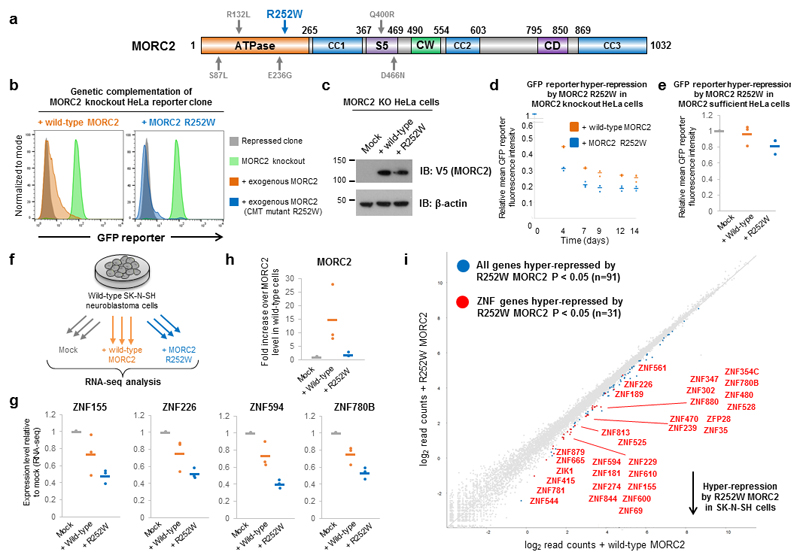
Dominant mutations in the MORC2 gene have recently been shown to cause axonal Charcot-Marie-Tooth (CMT) disease, but the cellular function of MORC2 is poorly understood. Here, through a genome-wide CRISPR-Cas9-mediated forward genetic screen, we identified MORC2 as an essential gene required for epigenetic silencing by the HUSH complex. HUSH recruits MORC2 to target sites in heterochromatin. We exploited a new method, differential viral accessibility (DIVA), to show that loss of MORC2 results in chromatin decompaction at these target loci, which is concomitant with a loss of H3K9me3 deposition and transcriptional derepression. The ATPase activity of MORC2 is critical for HUSH-mediated silencing, and the most common alteration affecting the ATPase domain in CMT patients (p.Arg252Trp) hyperactivates HUSH-mediated repression in neuronal cells. These data define a critical role for MORC2 in epigenetic silencing by the HUSH complex and provide a mechanistic basis underpinning the role of MORC2 mutations in CMT disease.
Conflict of interest statement
The authors declare that no competing financial interests exist.
Figures
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
