

Between-region genetic divergence reflects the mode and tempo of tumor evolution
- PMID: 28581503
- PMCID: PMC5643198
- DOI: 10.1038/ng.3891
Between-region genetic divergence reflects the mode and tempo of tumor evolution
Abstract
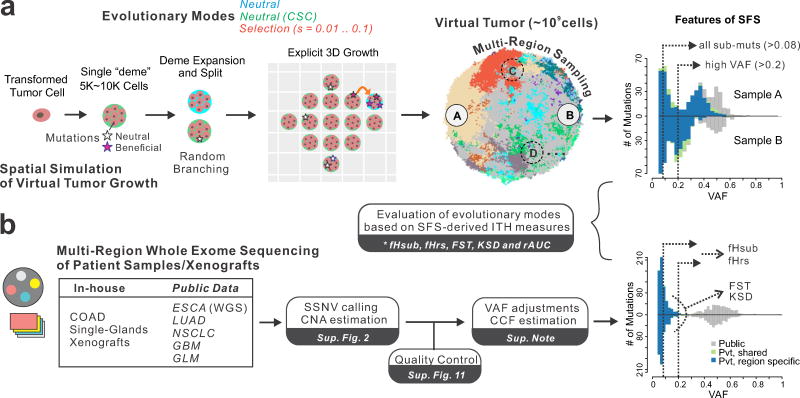
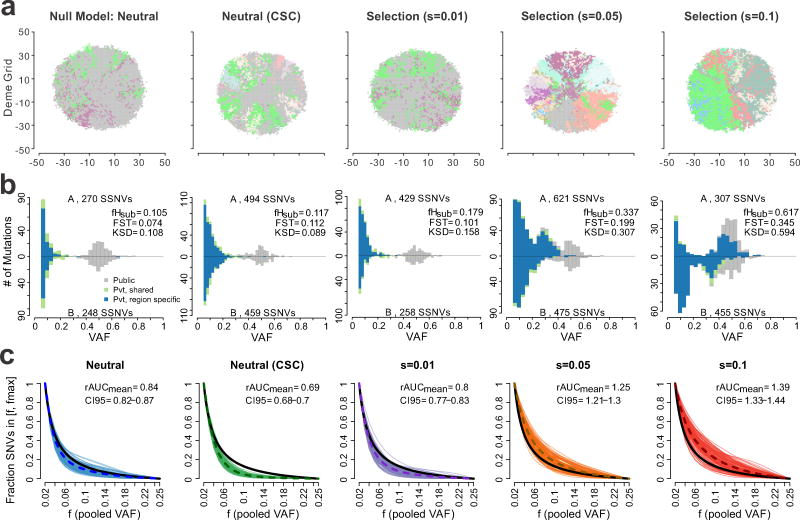
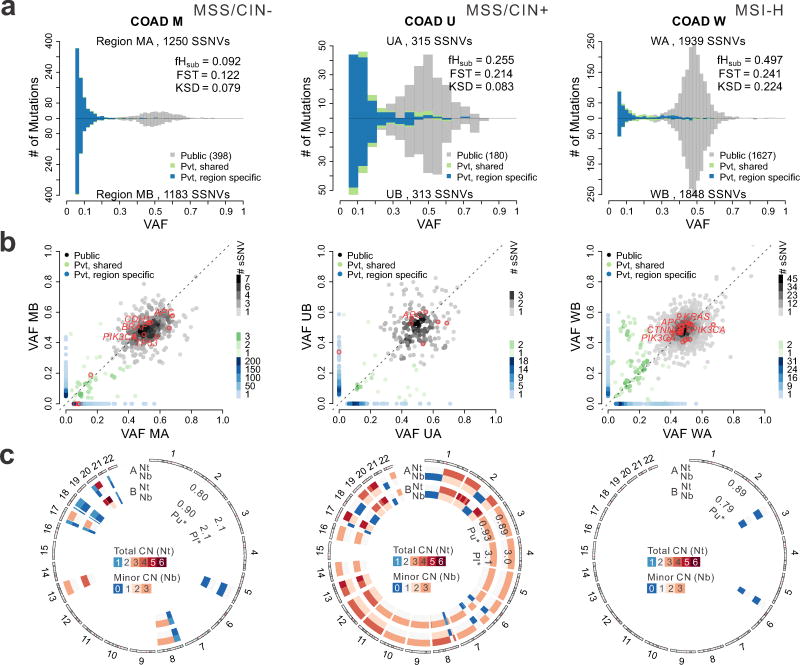
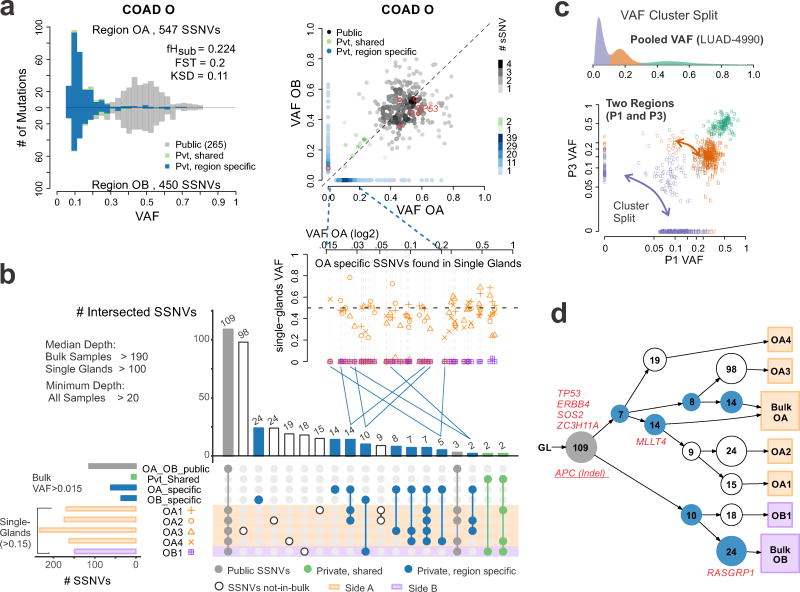
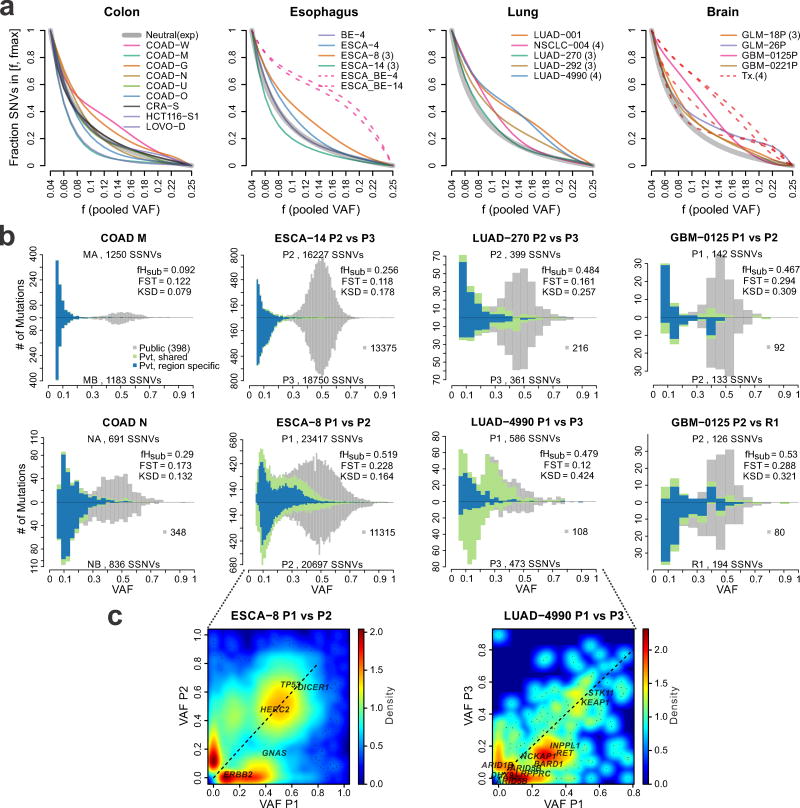
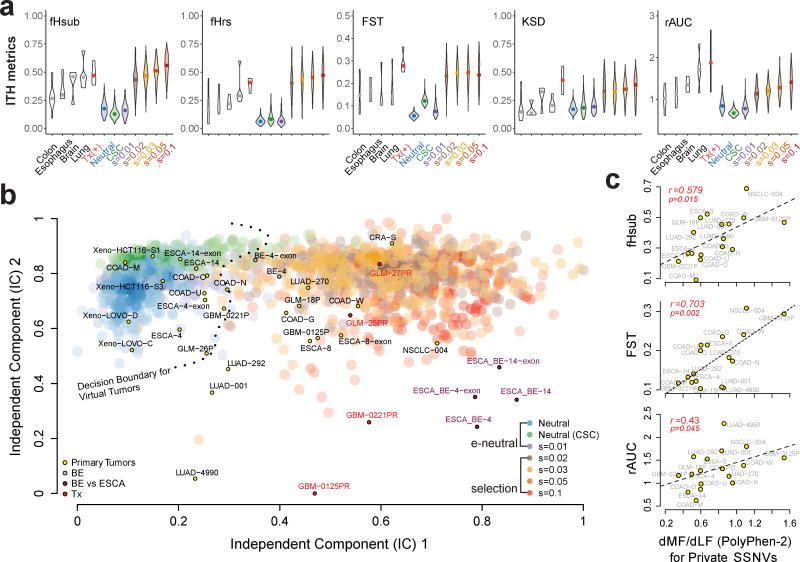
Given the implications of tumor dynamics for precision medicine, there is a need to systematically characterize the mode of evolution across diverse solid tumor types. In particular, methods to infer the role of natural selection within established human tumors are lacking. By simulating spatial tumor growth under different evolutionary modes and examining patterns of between-region subclonal genetic divergence from multiregion sequencing (MRS) data, we demonstrate that it is feasible to distinguish tumors driven by strong positive subclonal selection from those evolving neutrally or under weak selection, as the latter fail to dramatically alter subclonal composition. We developed a classifier based on measures of between-region subclonal genetic divergence and projected patient data into model space, finding different modes of evolution both within and between solid tumor types. Our findings have broad implications for how human tumors progress, how they accumulate intratumoral heterogeneity, and ultimately how they may be more effectively treated.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
