

A comprehensive assessment of RNA-seq protocols for degraded and low-quantity samples
- PMID: 28583074
- PMCID: PMC5460543
- DOI: 10.1186/s12864-017-3827-y
A comprehensive assessment of RNA-seq protocols for degraded and low-quantity samples
Abstract
Background: RNA-sequencing (RNA-seq) has emerged as one of the most sensitive tool for gene expression analysis. Among the library preparation methods available, the standard poly(A) + enrichment provides a comprehensive, detailed, and accurate view of polyadenylated RNAs. However, on samples of suboptimal quality ribosomal RNA depletion and exon capture methods have recently been reported as better alternatives.
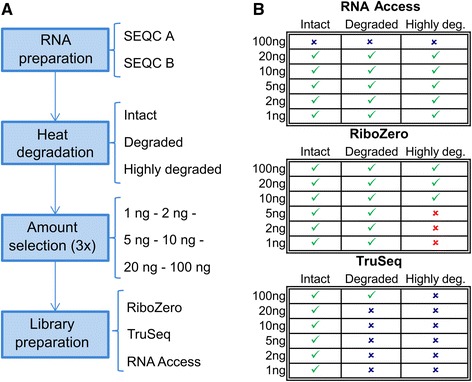
Methods: We compared for the first time three commercial Illumina library preparation kits (TruSeq Stranded mRNA, TruSeq Ribo-Zero rRNA Removal, and TruSeq RNA Access) as representatives of these three different approaches using well-established human reference RNA samples from the MAQC/SEQC consortium on a wide range of input amounts (from 100 ng down to 1 ng) and degradation levels (intact, degraded, and highly degraded).
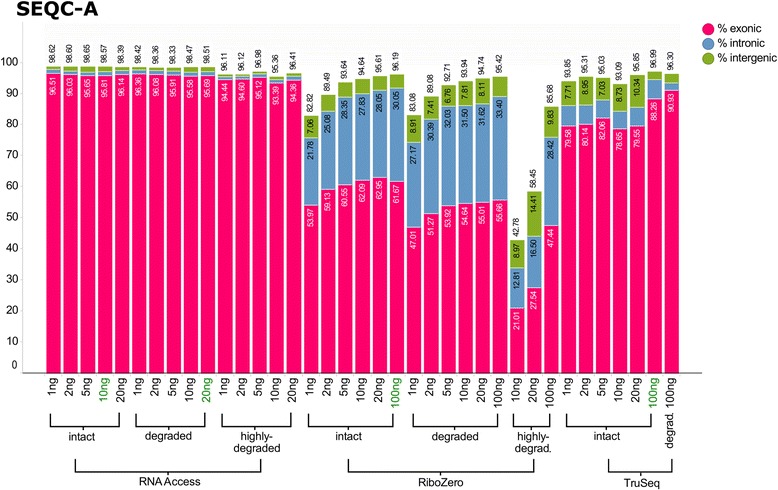
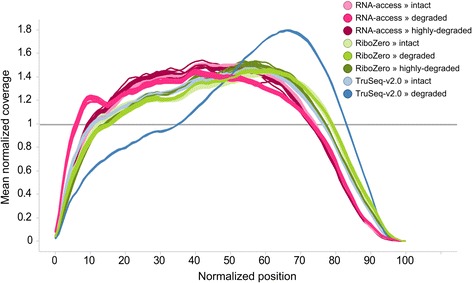
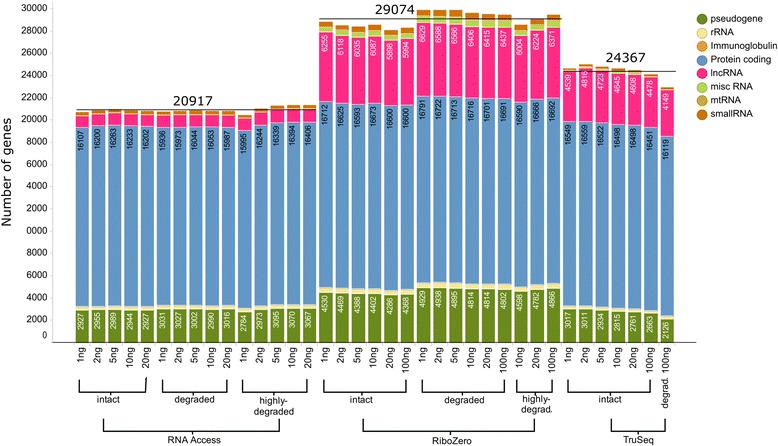
Results: We assessed the accuracy of the generated expression values by comparison to gold standard TaqMan qPCR measurements and gained unprecedented insight into the limits of applicability in terms of input quantity and sample quality of each protocol. We found that each protocol generates highly reproducible results (R 2 > 0.92) on intact RNA samples down to input amounts of 10 ng. For degraded RNA samples, Ribo-Zero showed clear performance advantages over the other two protocols as it generated more accurate and better reproducible gene expression results even at very low input amounts such as 1 ng and 2 ng. For highly degraded RNA samples, RNA Access performed best generating reliable data down to 5 ng input.
Conclusions: We found that the ribosomal RNA depletion protocol from Illumina works very well at amounts far below recommendation and over a good range of intact and degraded material. We also infer that the exome-capture protocol (RNA Access, Illumina) performs better than other methods on highly degraded and low amount samples.
Keywords: Benchmarking; Differential expression; Expression profiling; Low quality; Low quantity; RNA-sequencing.
Figures
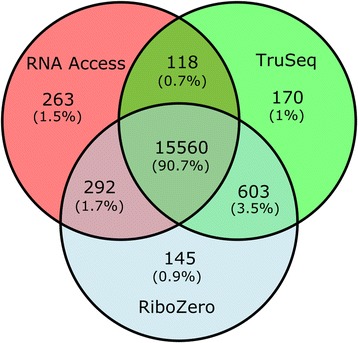
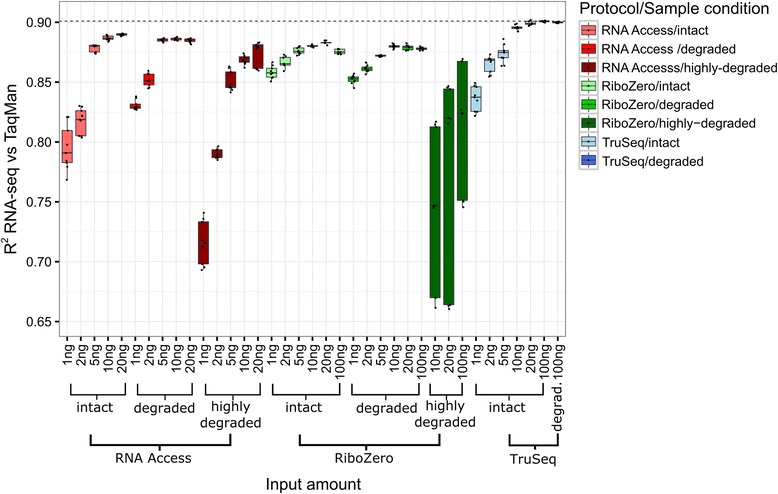
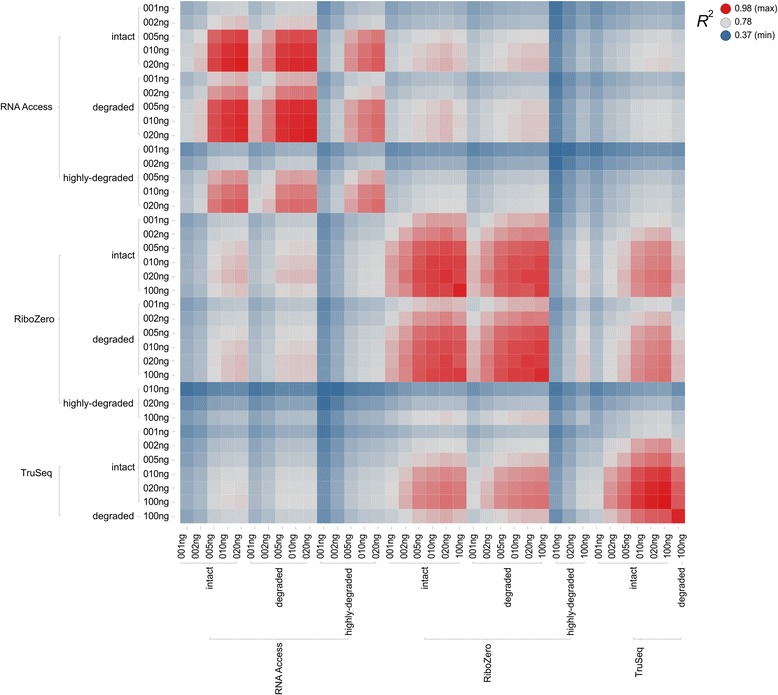
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
