

Noise Trauma-Induced Behavioral Gap Detection Deficits Correlate with Reorganization of Excitatory and Inhibitory Local Circuits in the Inferior Colliculus and Are Prevented by Acoustic Enrichment
- PMID: 28583912
- PMCID: PMC5490066
- DOI: 10.1523/JNEUROSCI.0602-17.2017
Noise Trauma-Induced Behavioral Gap Detection Deficits Correlate with Reorganization of Excitatory and Inhibitory Local Circuits in the Inferior Colliculus and Are Prevented by Acoustic Enrichment
Abstract
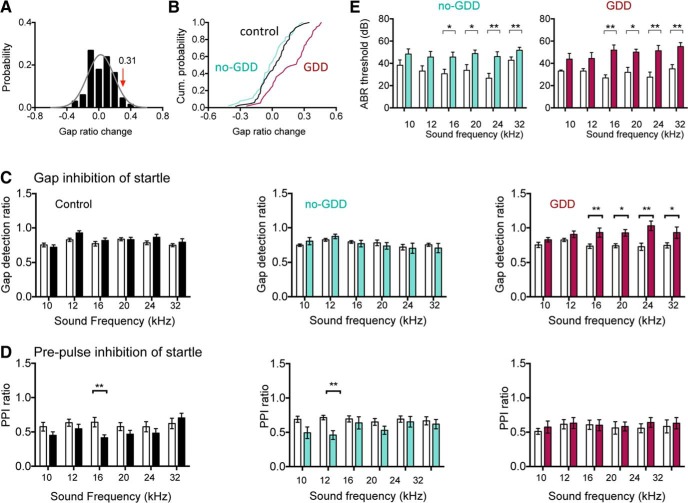
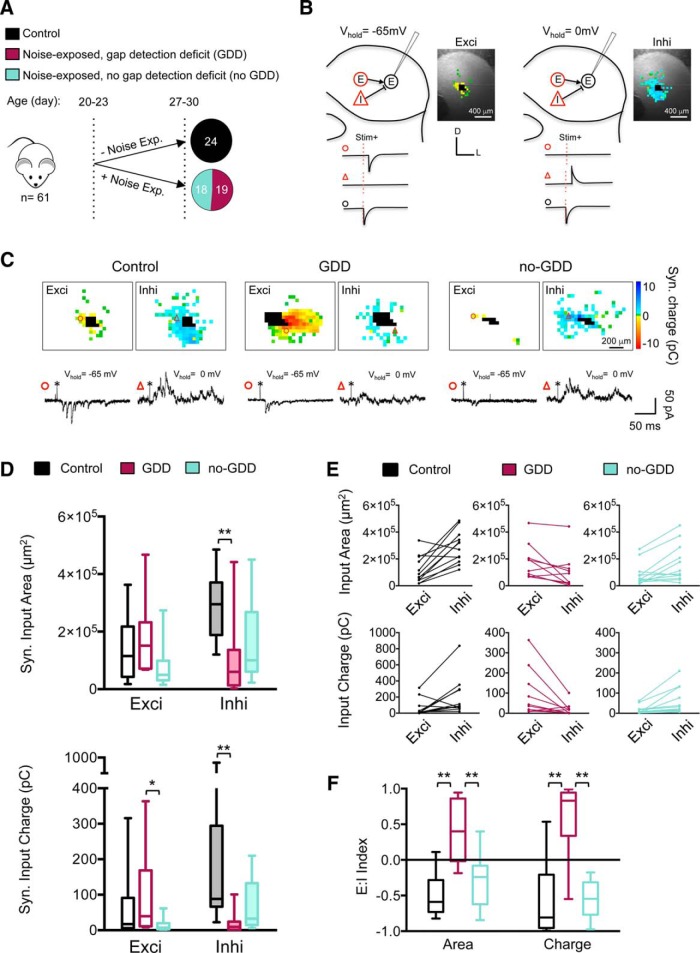
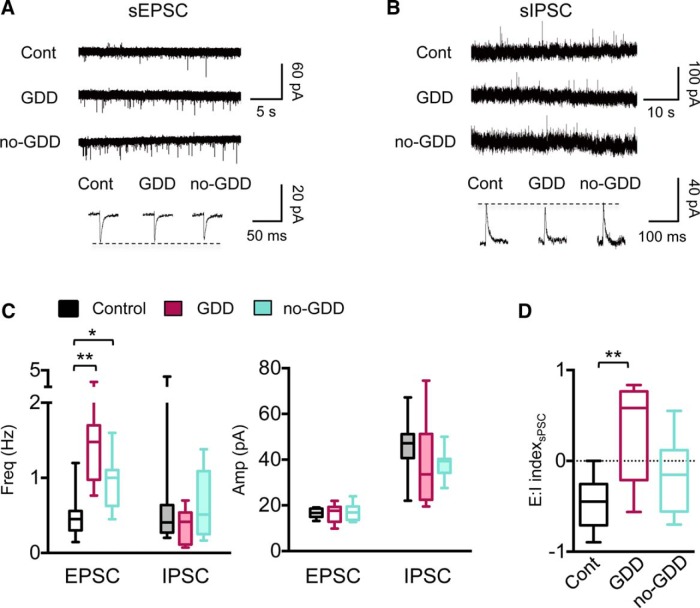
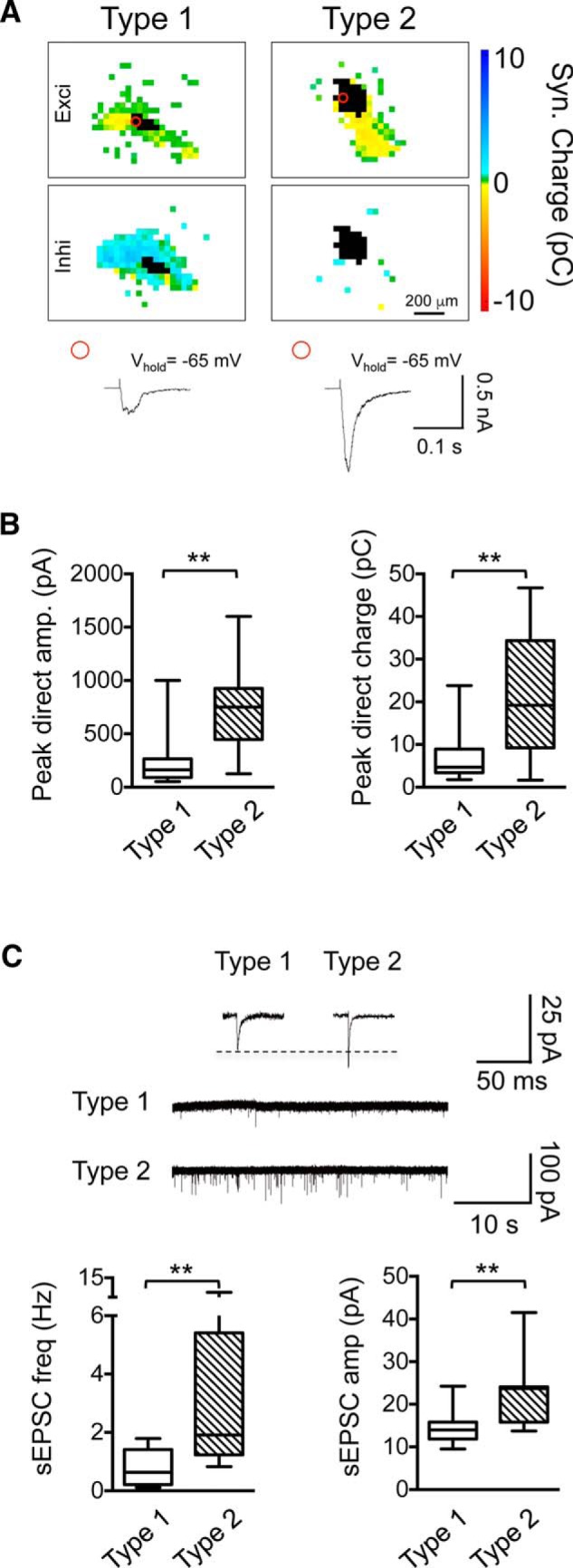
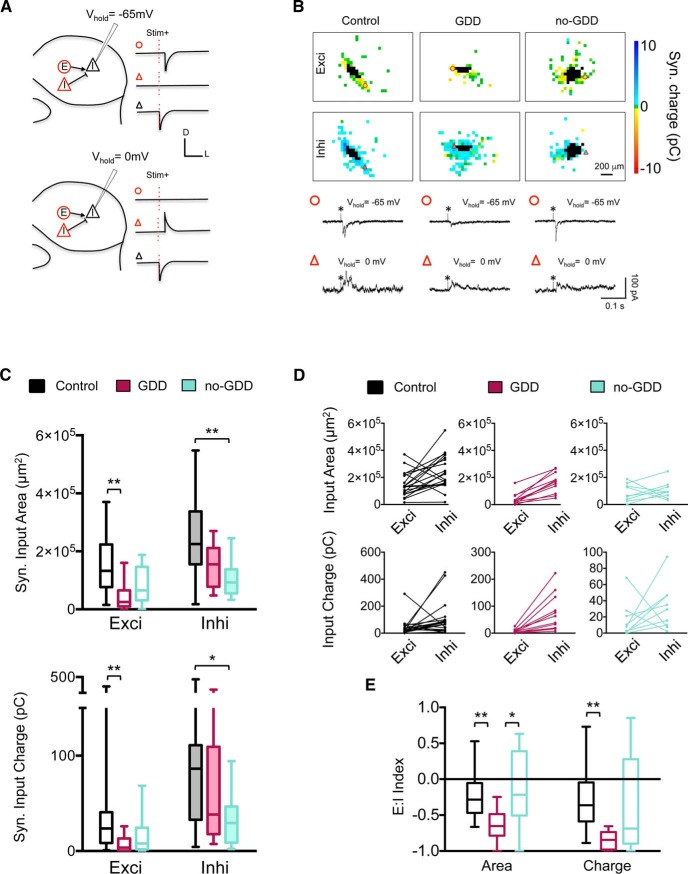
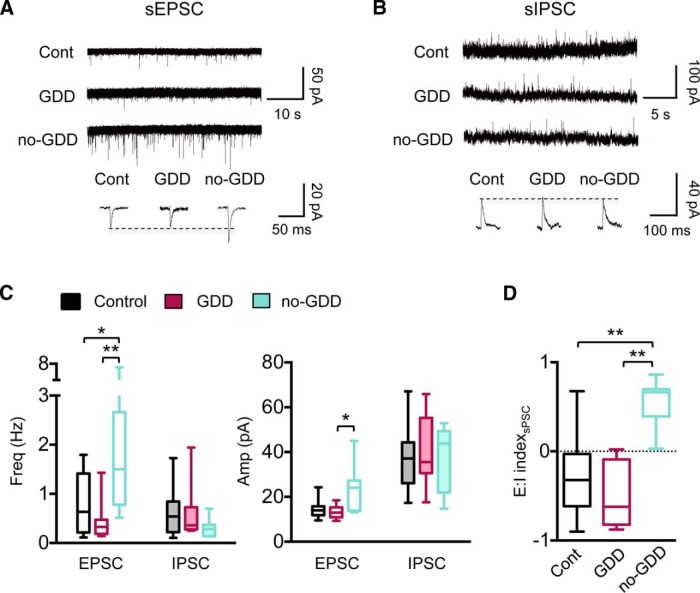
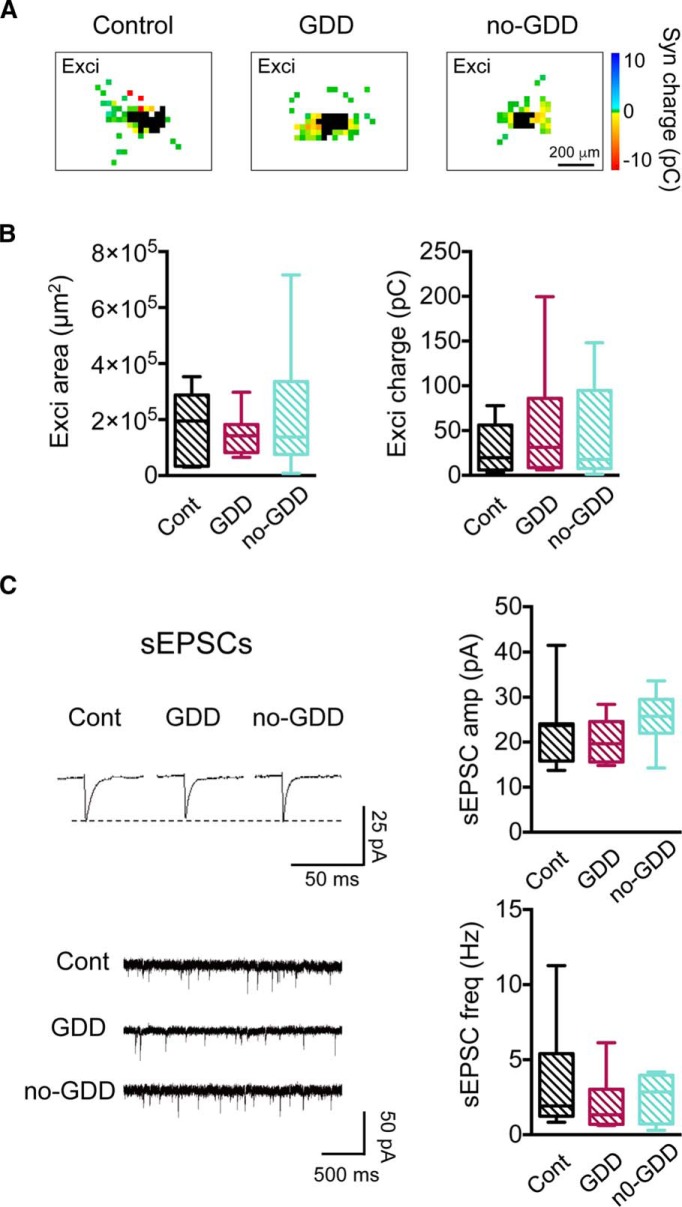
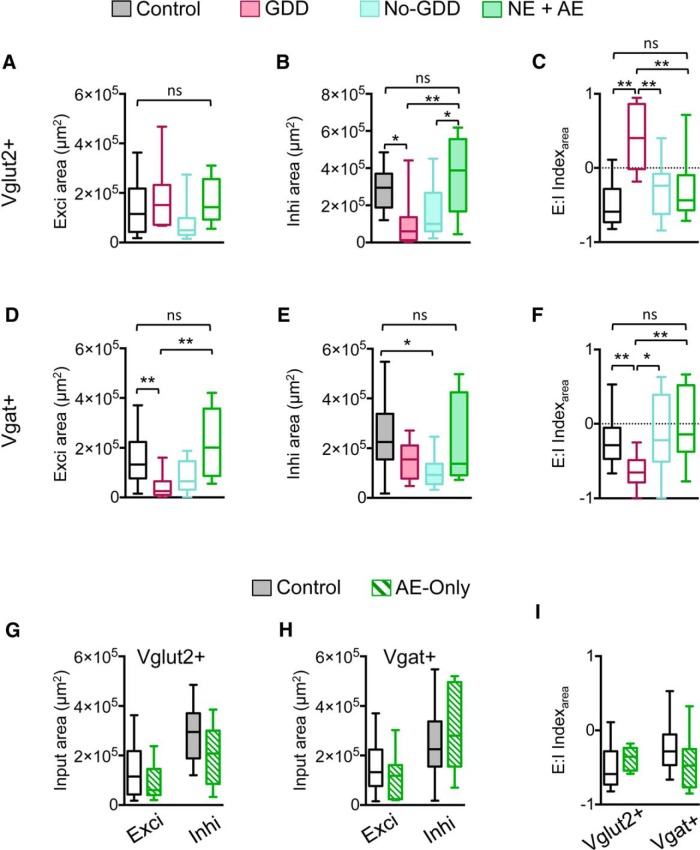
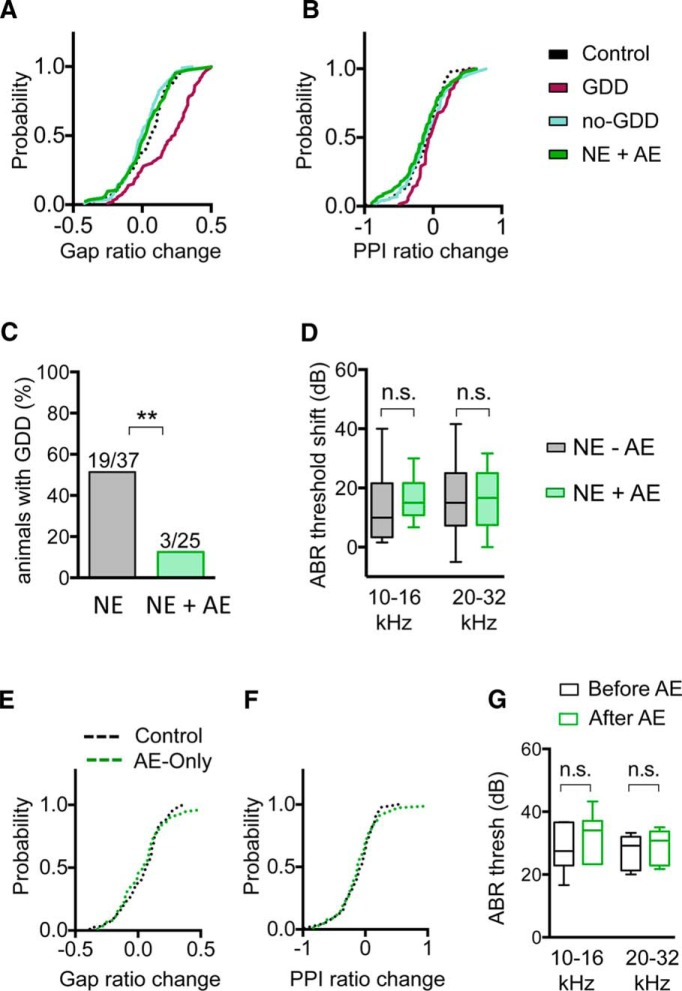
Hearing loss leads to a host of cellular and synaptic changes in auditory brain areas that are thought to give rise to auditory perception deficits such as temporal processing impairments, hyperacusis, and tinnitus. However, little is known about possible changes in synaptic circuit connectivity that may underlie these hearing deficits. Here, we show that mild hearing loss as a result of brief noise exposure leads to a pronounced reorganization of local excitatory and inhibitory circuits in the mouse inferior colliculus. The exact nature of these reorganizations correlated with the presence or absence of the animals' impairments in detecting brief sound gaps, a commonly used behavioral sign for tinnitus in animal models. Mice with gap detection deficits (GDDs) showed a shift in the balance of synaptic excitation and inhibition that was present in both glutamatergic and GABAergic neurons, whereas mice without GDDs showed stable excitation-inhibition balances. Acoustic enrichment (AE) with moderate intensity, pulsed white noise immediately after noise trauma prevented both circuit reorganization and GDDs, raising the possibility of using AE immediately after cochlear damage to prevent or alleviate the emergence of central auditory processing deficits.SIGNIFICANCE STATEMENT Noise overexposure is a major cause of central auditory processing disorders, including tinnitus, yet the changes in synaptic connectivity underlying these disorders remain poorly understood. Here, we find that brief noise overexposure leads to distinct reorganizations of excitatory and inhibitory synaptic inputs onto glutamatergic and GABAergic neurons and that the nature of these reorganizations correlates with animals' impairments in detecting brief sound gaps, which is often considered a sign of tinnitus. Acoustic enrichment immediately after noise trauma prevents circuit reorganizations and gap detection deficits, highlighting the potential for using sound therapy soon after cochlear damage to prevent the development of central processing deficits.
Keywords: midbrain; noise trauma; tinnitus; uncaging.
Copyright © 2017 the authors 0270-6474/17/376314-17$15.00/0.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
Similar articles
-
The dissimilar time course of temporary threshold shifts and reduction of inhibition in the inferior colliculus following intense sound exposure.Hear Res. 2014 Jun;312:38-47. doi: 10.1016/j.heares.2014.03.004. Epub 2014 Mar 18. Hear Res. 2014. PMID: 24650953
-
The immediate effects of acoustic trauma on excitation and inhibition in the inferior colliculus: A Wiener-kernel analysis.Hear Res. 2016 Jan;331:47-56. doi: 10.1016/j.heares.2015.10.007. Epub 2015 Oct 30. Hear Res. 2016. PMID: 26523371
-
Hyperexcitability of inferior colliculus and acoustic startle reflex with age-related hearing loss.Hear Res. 2017 Jul;350:32-42. doi: 10.1016/j.heares.2017.03.011. Epub 2017 Mar 27. Hear Res. 2017. PMID: 28431308 Free PMC article.
-
Cross-modal interactions of auditory and somatic inputs in the brainstem and midbrain and their imbalance in tinnitus and deafness.Am J Audiol. 2008 Dec;17(2):S193-209. doi: 10.1044/1059-0889(2008/07-0045). Am J Audiol. 2008. PMID: 19056923 Free PMC article. Review.
-
Tinnitus: Maladaptive auditory-somatosensory plasticity.Hear Res. 2016 Apr;334:20-9. doi: 10.1016/j.heares.2015.06.005. Epub 2015 Jun 12. Hear Res. 2016. PMID: 26074307 Free PMC article. Review.
Cited by
-
The Relative Contribution of Cochlear Synaptopathy and Reduced Inhibition to Age-Related Hearing Impairment for People With Normal Audiograms.Trends Hear. 2023 Jan-Dec;27:23312165231213191. doi: 10.1177/23312165231213191. Trends Hear. 2023. PMID: 37956654 Free PMC article.
-
Prolonged Exposure of CBA/Ca Mice to Moderately Loud Noise Can Cause Cochlear Synaptopathy but Not Tinnitus or Hyperacusis as Assessed With the Acoustic Startle Reflex.Trends Hear. 2018 Jan-Dec;22:2331216518758109. doi: 10.1177/2331216518758109. Trends Hear. 2018. PMID: 29532738 Free PMC article.
-
Neuropeptide Y signaling regulates recurrent excitation in the auditory midbrain.bioRxiv [Preprint]. 2023 May 17:2023.05.16.540954. doi: 10.1101/2023.05.16.540954. bioRxiv. 2023. Update in: J Neurosci. 2023 Nov 8;43(45):7626-7641. doi: 10.1523/JNEUROSCI.0900-23.2023. PMID: 37292904 Free PMC article. Updated. Preprint.
-
Embryonic medial ganglionic eminence cells survive and integrate into the inferior colliculus of adult mice.Hear Res. 2022 Jul;420:108520. doi: 10.1016/j.heares.2022.108520. Epub 2022 May 16. Hear Res. 2022. PMID: 35617926 Free PMC article.
-
Hearing loss and brain plasticity: the hyperactivity phenomenon.Brain Struct Funct. 2021 Sep;226(7):2019-2039. doi: 10.1007/s00429-021-02313-9. Epub 2021 Jun 7. Brain Struct Funct. 2021. PMID: 34100151 Review.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous