

Autism spectrum disorder: neuropathology and animal models
- PMID: 28584888
- PMCID: PMC5693718
- DOI: 10.1007/s00401-017-1736-4
Autism spectrum disorder: neuropathology and animal models
Abstract
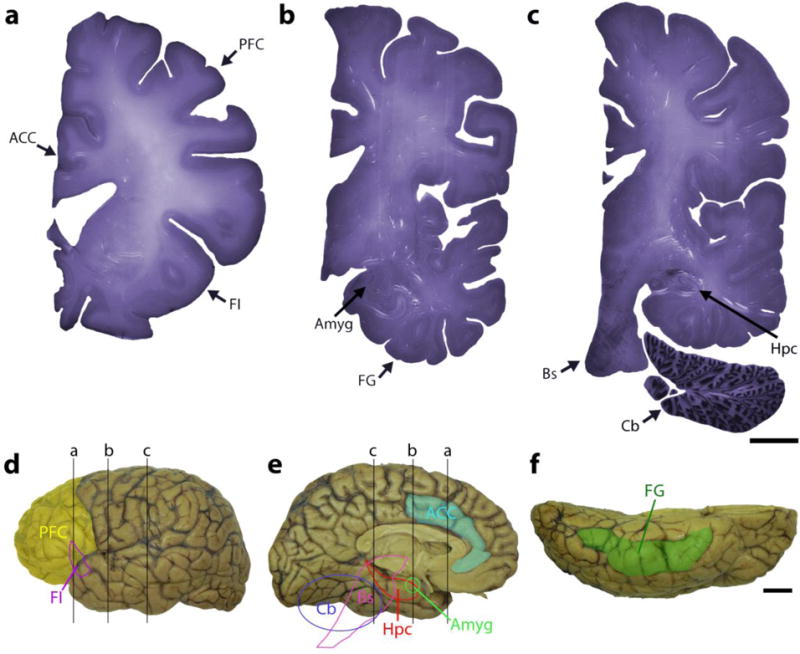
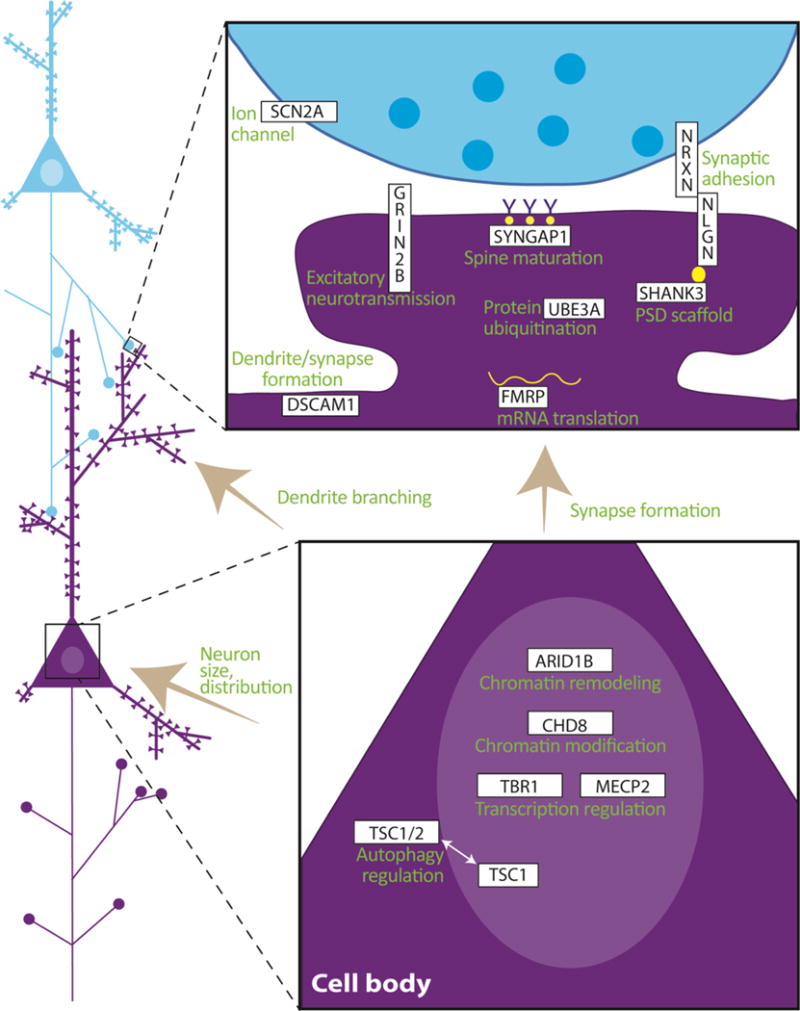
Autism spectrum disorder (ASD) has a major impact on the development and social integration of affected individuals and is the most heritable of psychiatric disorders. An increase in the incidence of ASD cases has prompted a surge in research efforts on the underlying neuropathologic processes. We present an overview of current findings in neuropathology studies of ASD using two investigational approaches, postmortem human brains and ASD animal models, and discuss the overlap, limitations, and significance of each. Postmortem examination of ASD brains has revealed global changes including disorganized gray and white matter, increased number of neurons, decreased volume of neuronal soma, and increased neuropil, the last reflecting changes in densities of dendritic spines, cerebral vasculature and glia. Both cortical and non-cortical areas show region-specific abnormalities in neuronal morphology and cytoarchitectural organization, with consistent findings reported from the prefrontal cortex, fusiform gyrus, frontoinsular cortex, cingulate cortex, hippocampus, amygdala, cerebellum and brainstem. The paucity of postmortem human studies linking neuropathology to the underlying etiology has been partly addressed using animal models to explore the impact of genetic and non-genetic factors clinically relevant for the ASD phenotype. Genetically modified models include those based on well-studied monogenic ASD genes (NLGN3, NLGN4, NRXN1, CNTNAP2, SHANK3, MECP2, FMR1, TSC1/2), emerging risk genes (CHD8, SCN2A, SYNGAP1, ARID1B, GRIN2B, DSCAM, TBR1), and copy number variants (15q11-q13 deletion, 15q13.3 microdeletion, 15q11-13 duplication, 16p11.2 deletion and duplication, 22q11.2 deletion). Models of idiopathic ASD include inbred rodent strains that mimic ASD behaviors as well as models developed by environmental interventions such as prenatal exposure to sodium valproate, maternal autoantibodies, and maternal immune activation. In addition to replicating some of the neuropathologic features seen in postmortem studies, a common finding in several animal models of ASD is altered density of dendritic spines, with the direction of the change depending on the specific genetic modification, age and brain region. Overall, postmortem neuropathologic studies with larger sample sizes representative of the various ASD risk genes and diverse clinical phenotypes are warranted to clarify putative etiopathogenic pathways further and to promote the emergence of clinically relevant diagnostic and therapeutic tools. In addition, as genetic alterations may render certain individuals more vulnerable to developing the pathological changes at the synapse underlying the behavioral manifestations of ASD, neuropathologic investigation using genetically modified animal models will help to improve our understanding of the disease mechanisms and enhance the development of targeted treatments.
Keywords: Autism spectrum disorder; Cerebral cortex; Genetically modified animal models; Idiopathic autism models; Neuronal morphology; Synapse.
Conflict of interest statement
Figures




References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
