

Global metabolic interaction network of the human gut microbiota for context-specific community-scale analysis
- PMID: 28585563
- PMCID: PMC5467172
- DOI: 10.1038/ncomms15393
Global metabolic interaction network of the human gut microbiota for context-specific community-scale analysis
Abstract
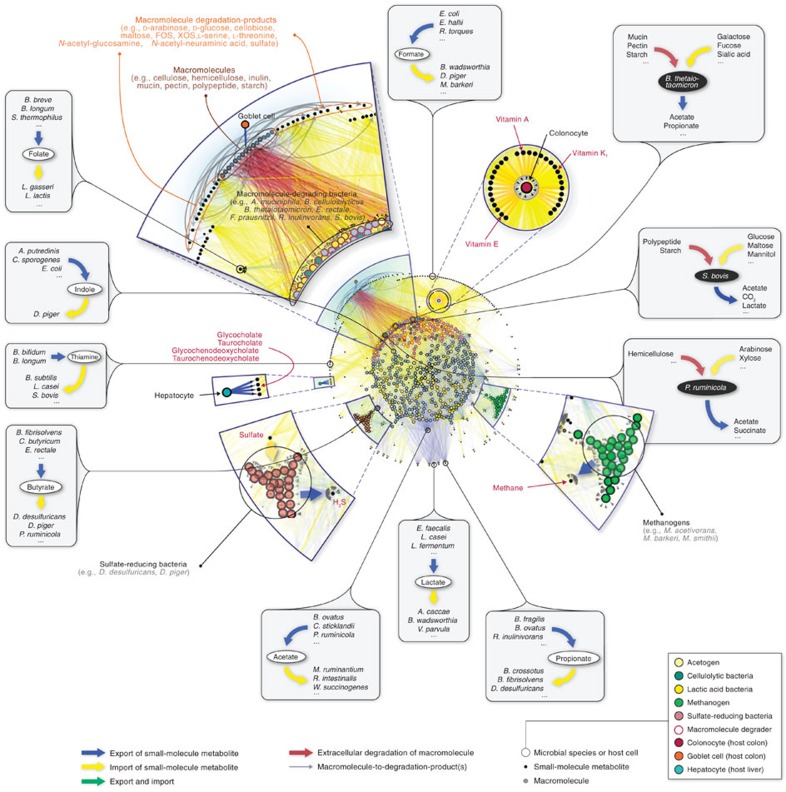
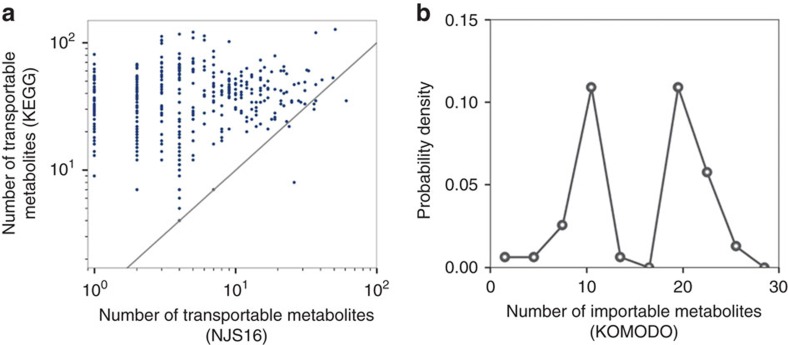
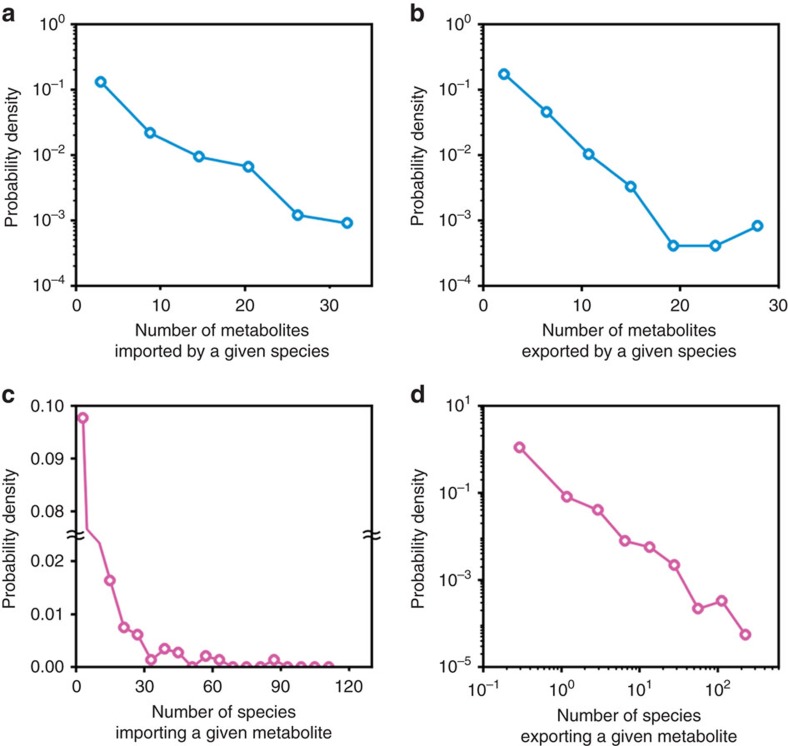
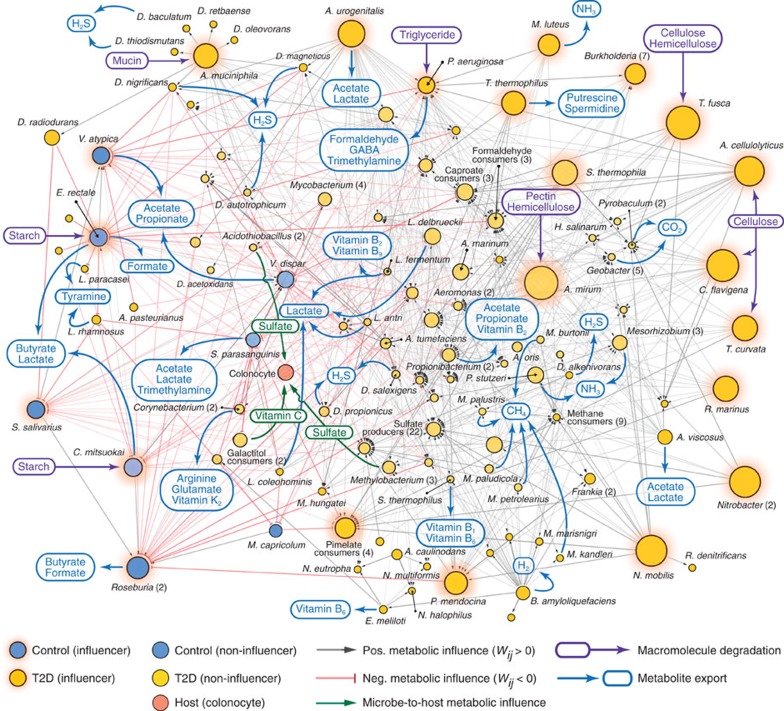
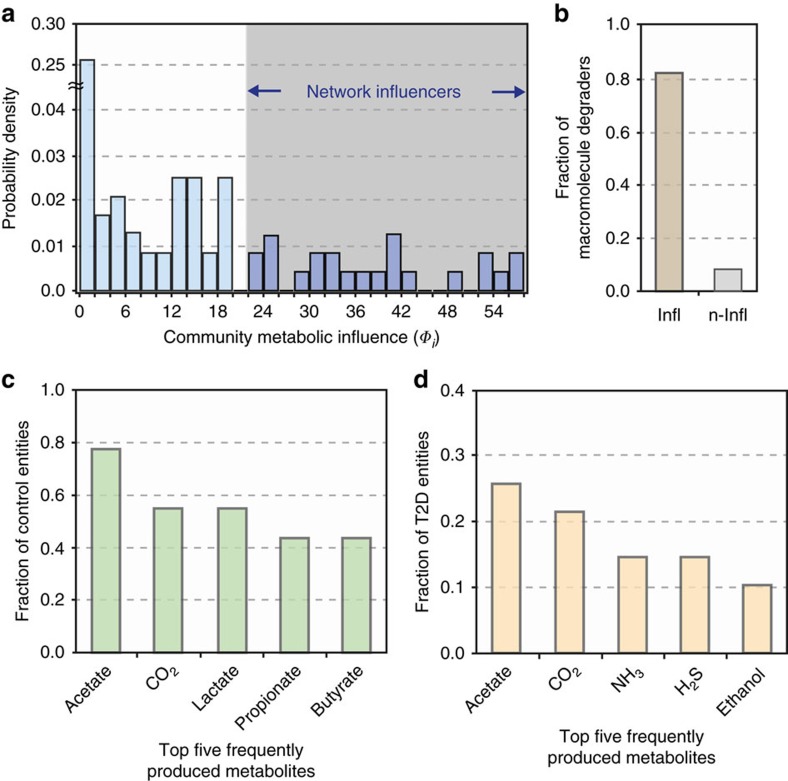
A system-level framework of complex microbe-microbe and host-microbe chemical cross-talk would help elucidate the role of our gut microbiota in health and disease. Here we report a literature-curated interspecies network of the human gut microbiota, called NJS16. This is an extensive data resource composed of ∼570 microbial species and 3 human cell types metabolically interacting through >4,400 small-molecule transport and macromolecule degradation events. Based on the contents of our network, we develop a mathematical approach to elucidate representative microbial and metabolic features of the gut microbial community in a given population, such as a disease cohort. Applying this strategy to microbiome data from type 2 diabetes patients reveals a context-specific infrastructure of the gut microbial ecosystem, core microbial entities with large metabolic influence, and frequently produced metabolic compounds that might indicate relevant community metabolic processes. Our network presents a foundation towards integrative investigations of community-scale microbial activities within the human gut.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
References
-
- Nowak M. A. Evolutionary Dynamics: exploring the Equations of Life Harvard Univ. Press (2006).
-
- Mackie R. I., White B. A. & Isaacson R. E. Gastrointestinal Microbiology Chapman & Hall (1997).
-
- Blaut M. Ecology and physiology of the intestinal tract. Curr. Top. Microbiol. Immunol. 358, 247–272 (2013). - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
