

Metabolic injury-induced NLRP3 inflammasome activation dampens phospholipid degradation
- PMID: 28588189
- PMCID: PMC5460122
- DOI: 10.1038/s41598-017-01994-9
Metabolic injury-induced NLRP3 inflammasome activation dampens phospholipid degradation
Abstract
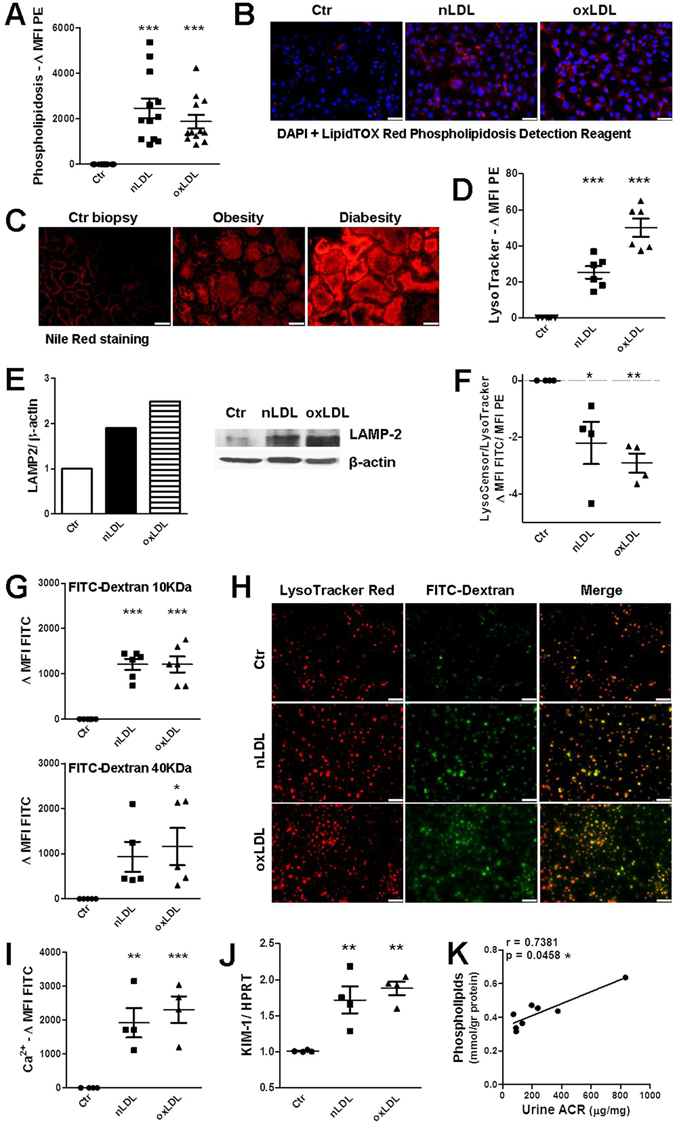
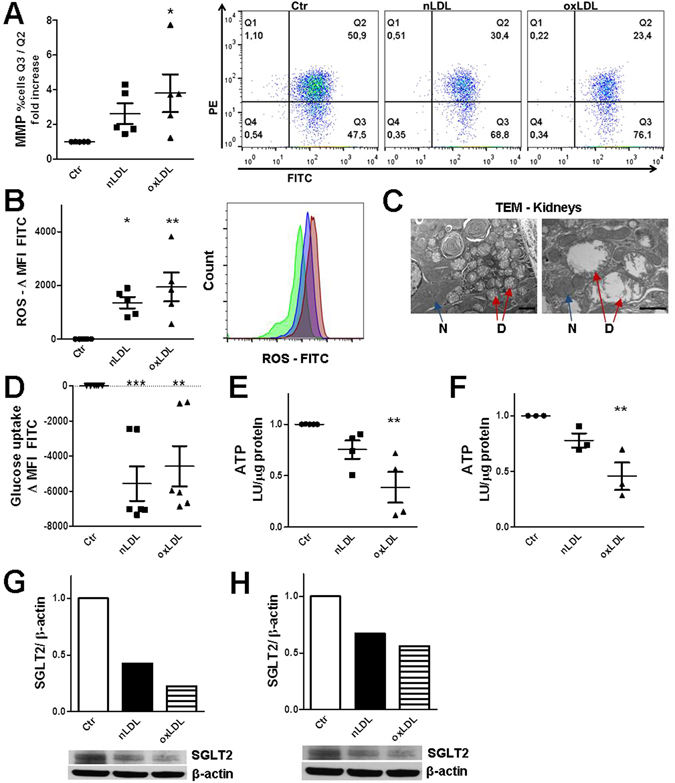
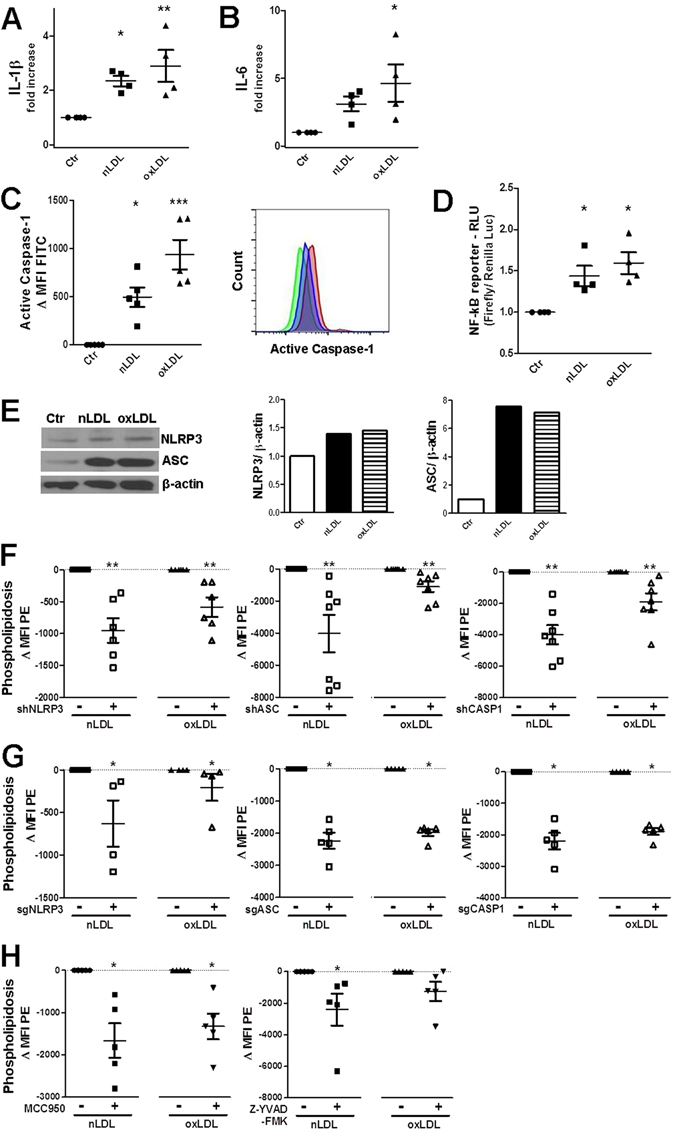
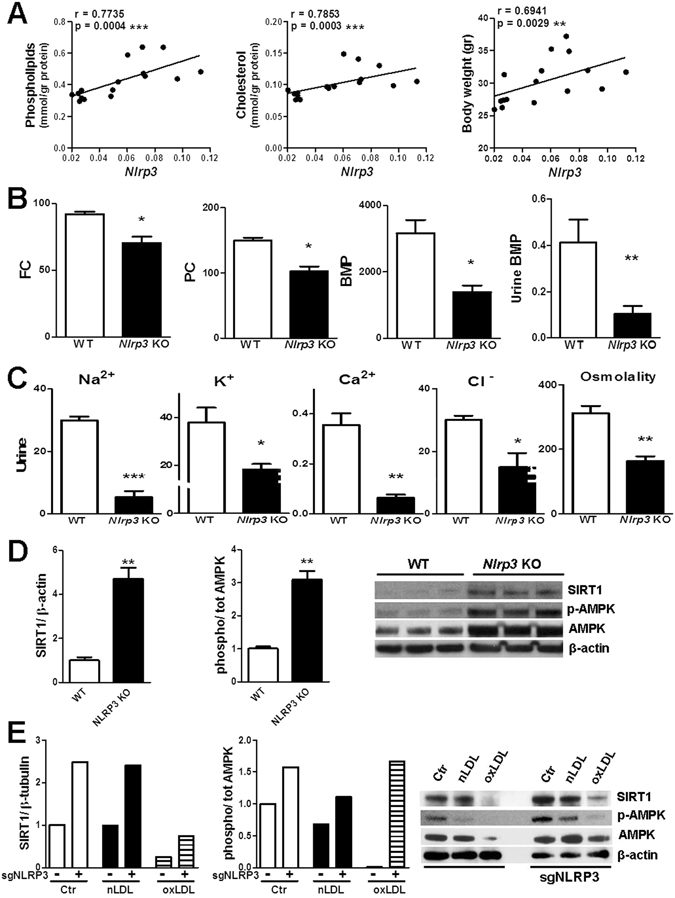
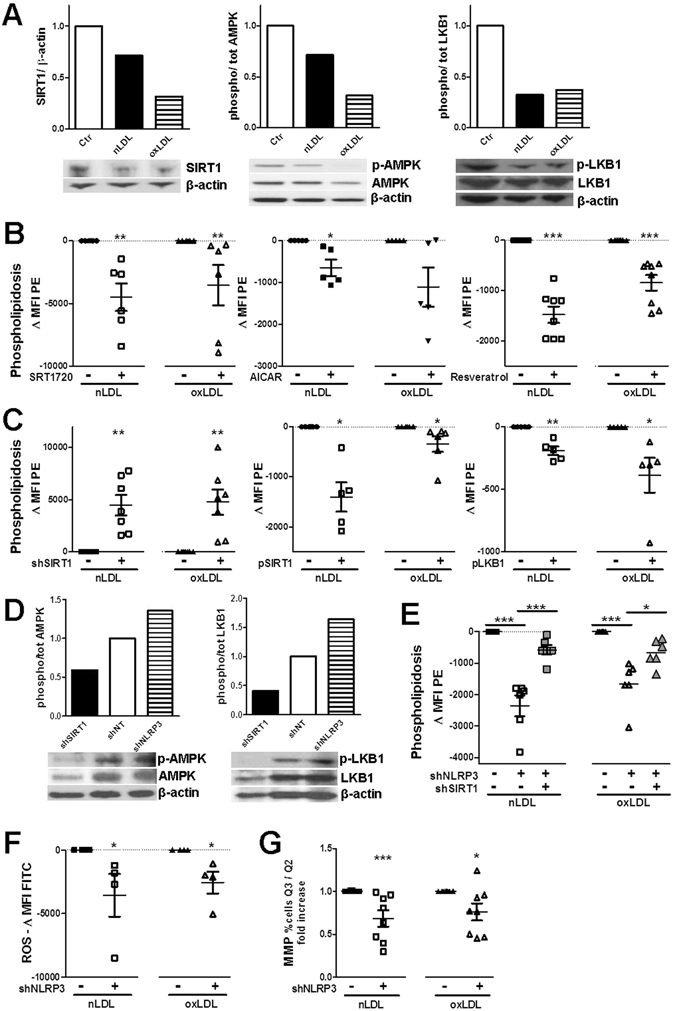
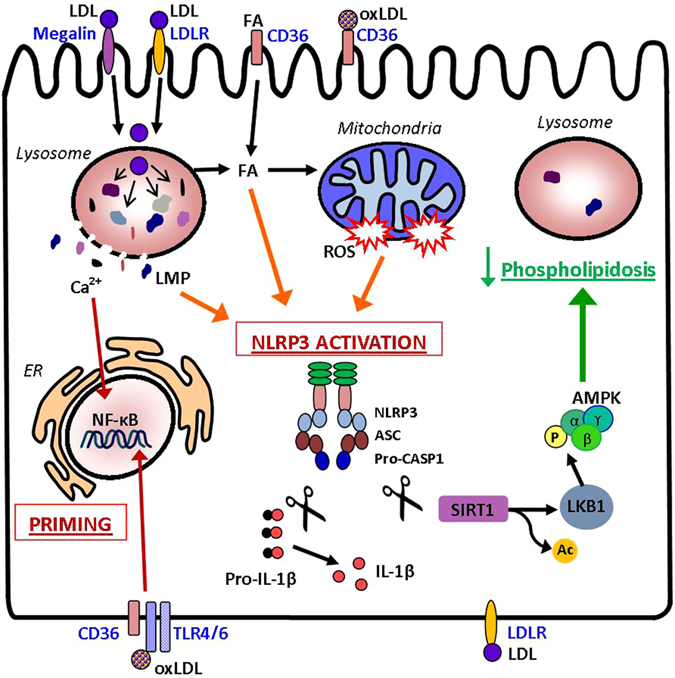
The collateral effects of obesity/metabolic syndrome include inflammation and renal function decline. As renal disease in obesity can occur independently of hypertension and diabetes, other yet undefined causal pathological pathways must be present. Our study elucidate novel pathological pathways of metabolic renal injury through LDL-induced lipotoxicity and metainflammation. Our in vitro and in vivo analysis revealed a direct lipotoxic effect of metabolic overloading on tubular renal cells through a multifaceted mechanism that includes intralysosomal lipid amassing, lysosomal dysfunction, oxidative stress, and tubular dysfunction. The combination of these endogenous metabolic injuries culminated in the activation of the innate immune NLRP3 inflammasome complex. By inhibiting the sirtuin-1/LKB1/AMPK pathway, NLRP3 inflammasome dampened lipid breakdown, thereby worsening the LDL-induced intratubular phospholipid accumulation. Consequently, the presence of NLRP3 exacerbated tubular oxidative stress, mitochondrial damage and malabsorption during overnutrition. Altogether, our data demonstrate a causal link between LDL and tubular damage and the creation of a vicious cycle of excessive nutrients-NLRP3 activation-catabolism inhibition during metabolic kidney injury. Hence, this study strongly highlights the importance of renal epithelium in lipid handling and recognizes the role of NLRP3 as a central hub in metainflammation and immunometabolism in parenchymal non-immune cells.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
