

RIP1 is a central signaling protein in regulation of TNF-α/TRAIL mediated apoptosis and necroptosis during Newcastle disease virus infection
- PMID: 28591723
- PMCID: PMC5522139
- DOI: 10.18632/oncotarget.17970
RIP1 is a central signaling protein in regulation of TNF-α/TRAIL mediated apoptosis and necroptosis during Newcastle disease virus infection
Abstract
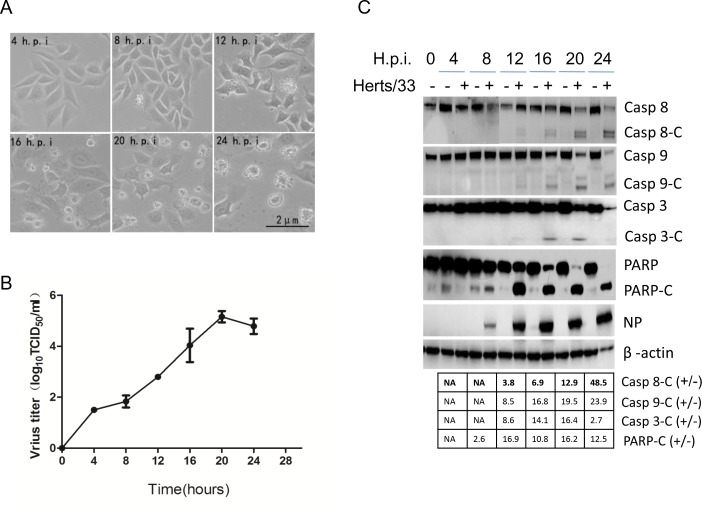
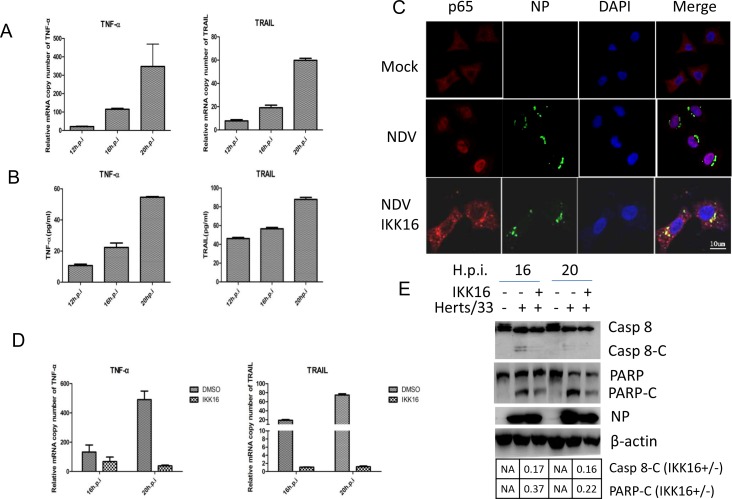
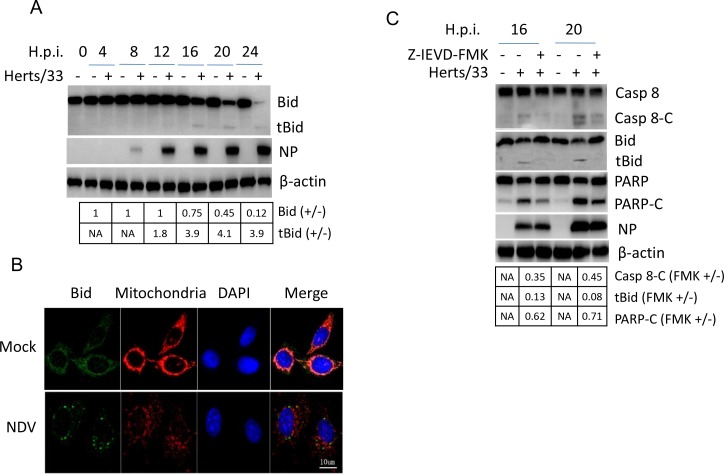
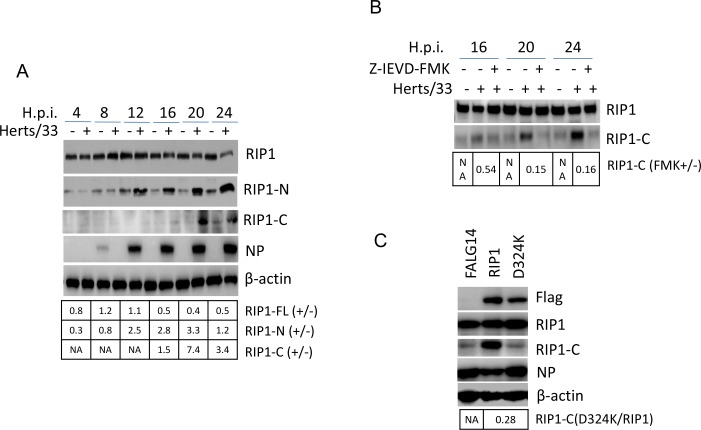
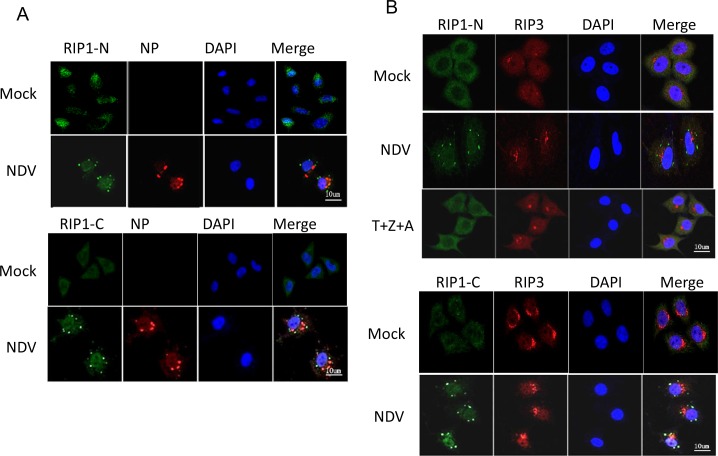
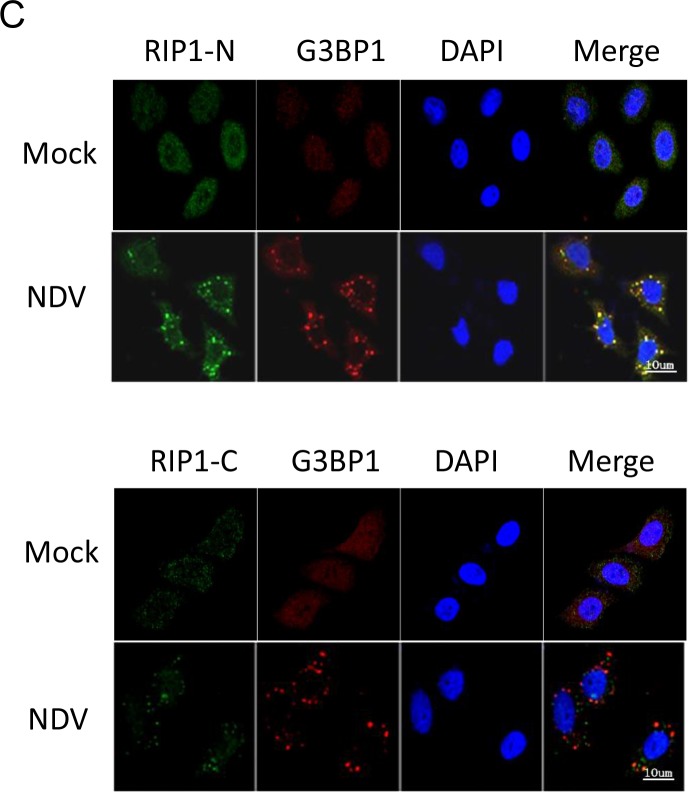
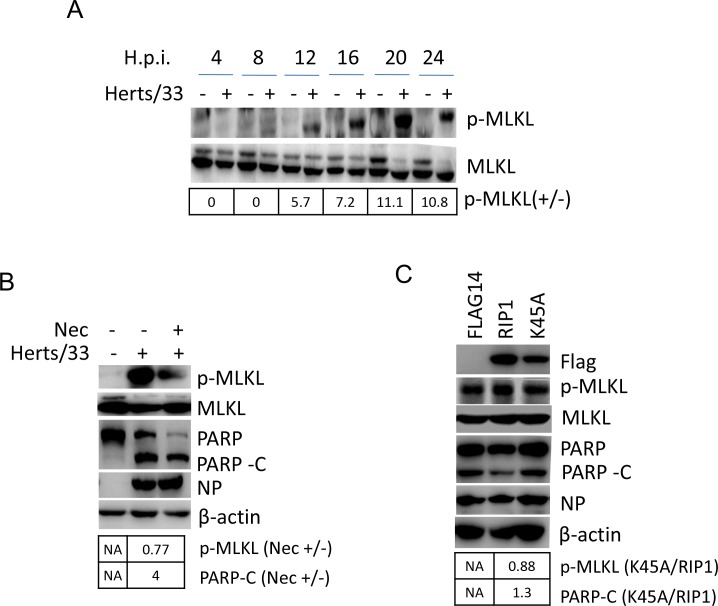
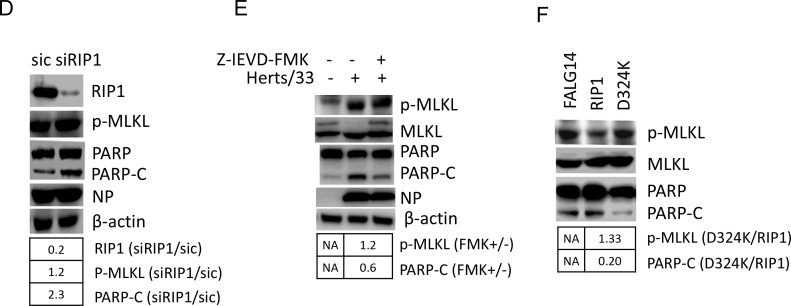
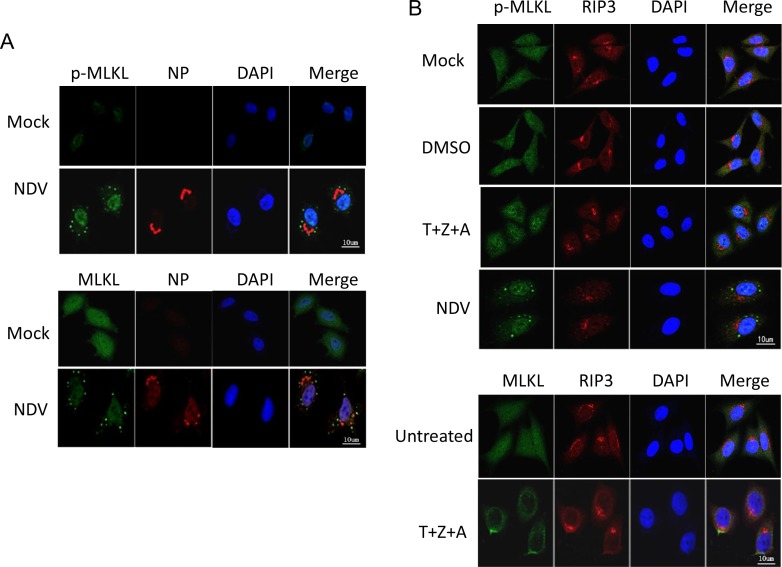
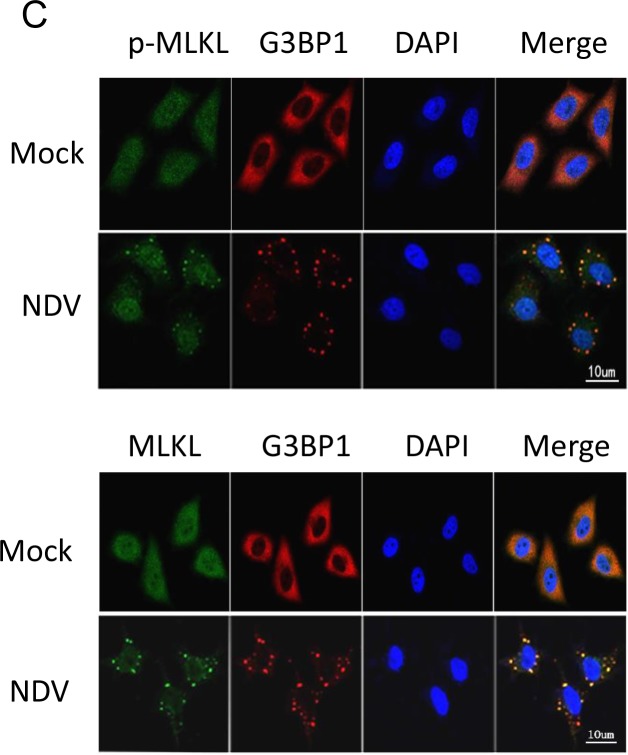
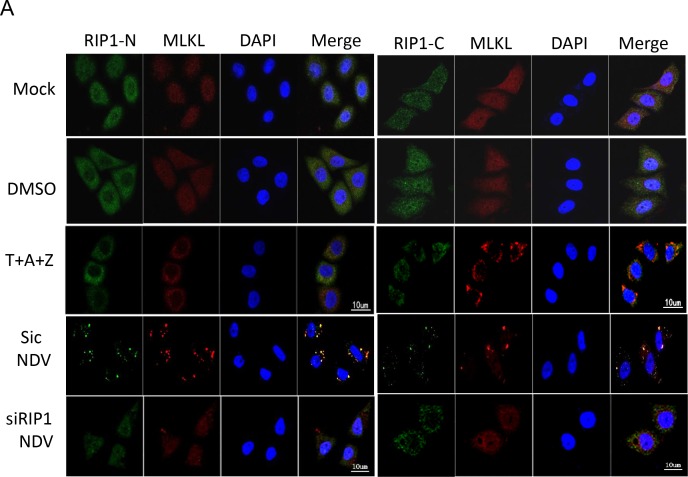
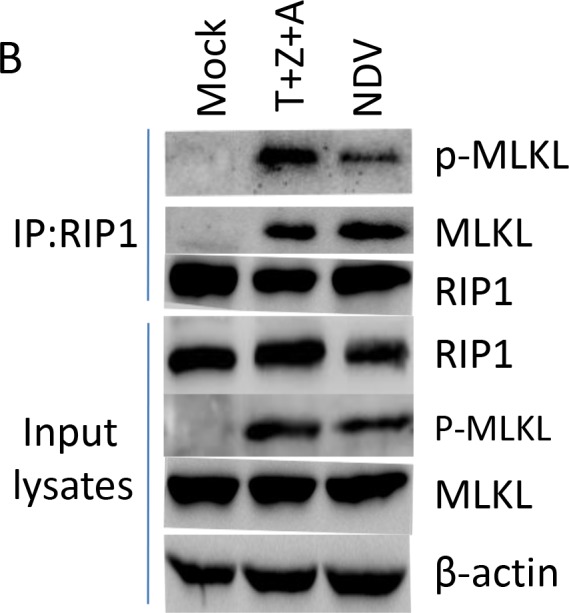
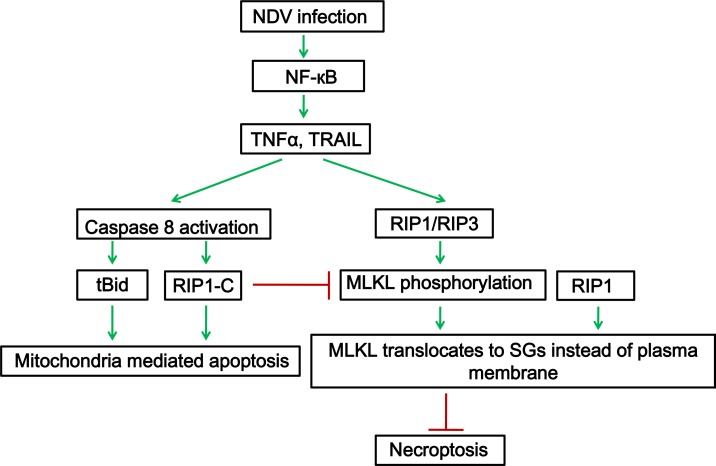
Newcastle disease virus (NDV) is an oncolytic virus which selectively replicates in tumor cells and exerts anti-tumor cytotoxic activity by promoting cell death. In this study, we focus on characterization of the underlying mechanisms of NDV-induced cell death in HeLa cells. We find that NDV Herts/33 strain triggers both extrinsic and intrinsic apoptosis at late infection times. The activation of NF-кB pathway and subsequent up-regulation of TNF-α/TRAIL initiates extrinsic apoptosis, leading to activation of caspase 8 and cleavage of Bid into tBid. tBid transmits the extrinsic apoptotic signals to mitochondria and mediates intrinsic apoptosis, which is hallmarked by cleavage of caspase 9. Moreover, RIP1 is cleaved into RIP1-N and RIP1-C at D324 by caspase 8, and this cleavage promotes apoptosis. Surprisingly, over expression of RIP1 reduces apoptosis and depletion of RIP1 promotes apoptosis, suggesting full length RIP1 is anti-apoptotic. Moreover, necroptosis hallmark protein MLKL is activated by phosphorylation at 12-24 h.p.i., and RIP1 regulates the level of phosphor-MLKL. Immunostaining shows that RIP1 aggregates to stress granules (SGs) at 8-24 h.p.i., and phosphor-MLKL is also recruited to SGs, instead of migrating to plasma membrane to exert its necrotic function. Immunoprecipitation study demonstrates that RIP1 bind to phosphor-MLKL, and depletion of RIP1 reduces the aggregation of MLKL to SGs, suggesting that RIP1 recruits MLKL to SGs. Altogether, NDV infection initiates extrinsic apoptosis via activation of NF-кB and secretion of TNF-α/TRAIL. Activation of caspase 8 by TNF-α/TRAIL and subsequent cleavage of Bid and RIP1 transmit the death signals to mitochondria. Meanwhile, virus subverts the host defensive necroptosis via recruiting phosphor-MLKL by RIP1 to SGs. Thus, RIP1 is a central signaling protein in regulation of apoptosis and necroptosis during NDV infection.
Keywords: NDV; RIP1; apoptosis; necroptosis.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
References
-
- Alexander DJ, Aldous EW, Fuller CM. The long view: a selective review of 40 years of Newcastle disease research. Avian Pathol. 2012;41:329–335. - PubMed
-
- Czegledi A, Ujvari D, Somogyi E, Wehmann E, Werner O, Lomniczi B. Third genome size category of avian paramyxovirus serotype 1 (Newcastle disease virus) and evolutionary implications. Virus Res. 2006;120:36–48. - PubMed
-
- Villar E, Barroso IM. Role of sialic acid-containing molecules in paramyxovirus entry into the host cell: a minireview. Glycoconj J. 2006;23:5–17. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
