

Severe congenital neutropenias
- PMID: 28593997
- PMCID: PMC5821468
- DOI: 10.1038/nrdp.2017.32
Severe congenital neutropenias
Abstract
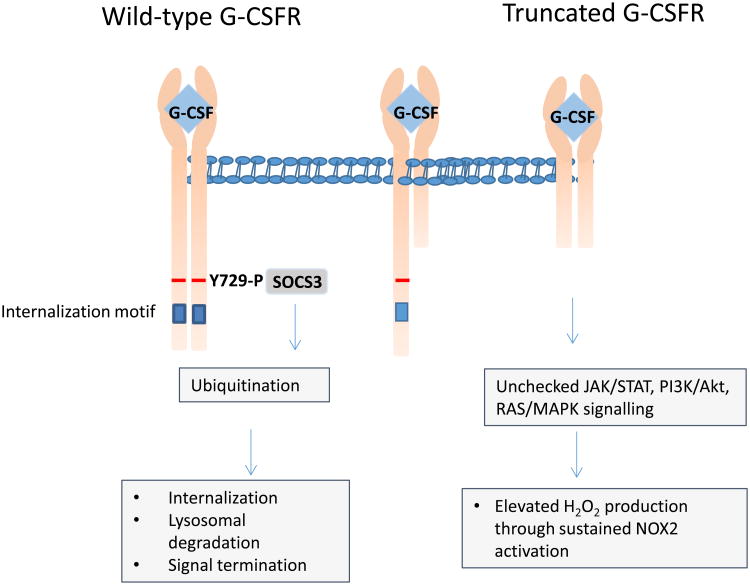
Severe congenital neutropenias are a heterogeneous group of rare haematological diseases characterized by impaired maturation of neutrophil granulocytes. Patients with severe congenital neutropenia are prone to recurrent, often life-threatening infections beginning in their first months of life. The most frequent pathogenic defects are autosomal dominant mutations in ELANE, which encodes neutrophil elastase, and autosomal recessive mutations in HAX1, whose product contributes to the activation of the granulocyte colony-stimulating factor (G-CSF) signalling pathway. The pathophysiological mechanisms of these conditions are the object of extensive research and are not fully understood. Furthermore, severe congenital neutropenias may predispose to myelodysplastic syndromes or acute myeloid leukaemia. Molecular events in the malignant progression include acquired mutations in CSF3R (encoding G-CSF receptor) and subsequently in other leukaemia-associated genes (such as RUNX1) in a majority of patients. Diagnosis is based on clinical manifestations, blood neutrophil count, bone marrow examination and genetic and immunological analyses. Daily subcutaneous G-CSF administration is the treatment of choice and leads to a substantial increase in blood neutrophil count, reduction of infections and drastic improvement of quality of life. Haematopoietic stem cell transplantation is the alternative treatment. Regular clinical assessments (including yearly bone marrow examinations) to monitor treatment course and detect chromosomal abnormalities (for example, monosomy 7 and trisomy 21) as well as somatic pre-leukaemic mutations are recommended.
Figures








References
-
- Welte K, Zeidler C, Dale DC. Severe congenital neutropenia. Semin Hematol. 2006;43:189–195. - PubMed
-
- Skokowa J, Germeshausen M, Zeidler C, Welte K. Severe congenital neutropenia: inheritance and pathophysiology. Curr Opin Hematol. 2007;14:22–28. - PubMed
-
- Donadieu J, Beaupain B, Mahlaoui N, Bellanne-Chantelot C. Epidemiology of congenital neutropenia. Hematol Oncol Clin North Am. 2013;27:1–17. - PubMed
-
- Carlsson G, et al. Incidence of severe congenital neutropenia in Sweden and risk of evolution to myelodysplastic syndrome/leukaemia. Br J Haematol. 2012;158:363–369. - PubMed
-
- Lebel A, Yacobovich J, Krasnov T, et al. Genetic analysis and clinical picture of severe congenital neutropenia in Israel. Pediatr Blood Cancer. 2015;62:103–108. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
