

Indoxyl sulfate (IS)-mediated immune dysfunction provokes endothelial damage in patients with end-stage renal disease (ESRD)
- PMID: 28596556
- PMCID: PMC5465082
- DOI: 10.1038/s41598-017-03130-z
Indoxyl sulfate (IS)-mediated immune dysfunction provokes endothelial damage in patients with end-stage renal disease (ESRD)
Abstract
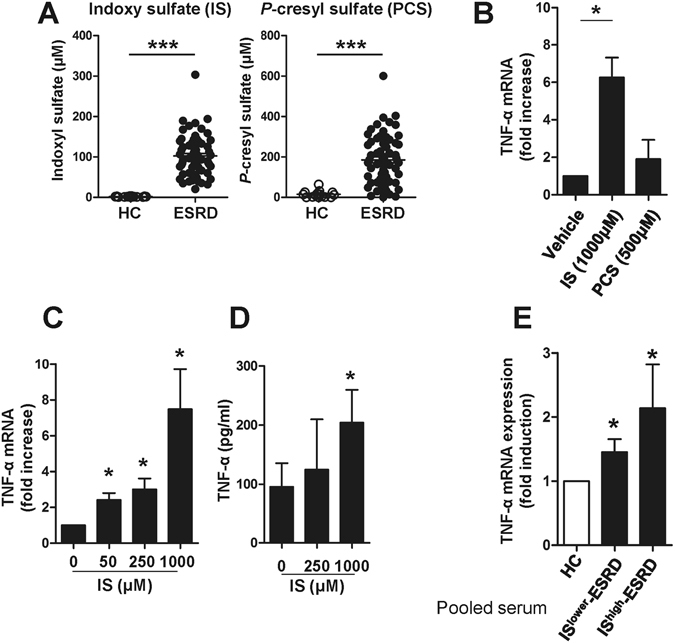
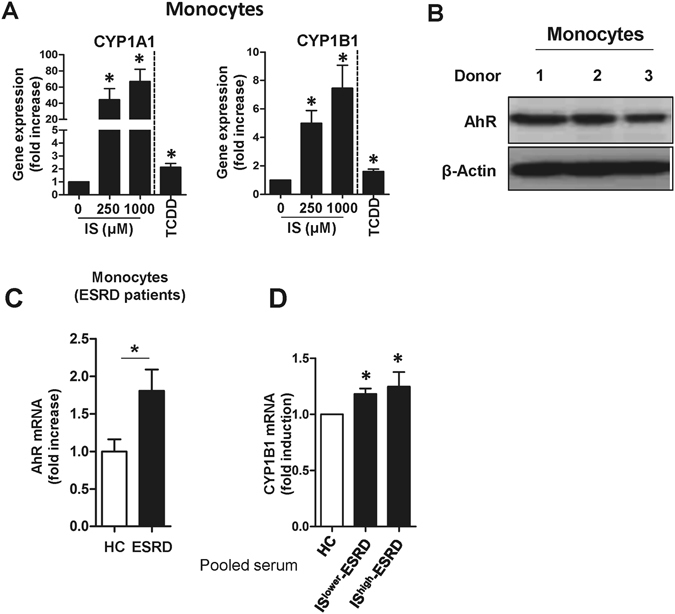
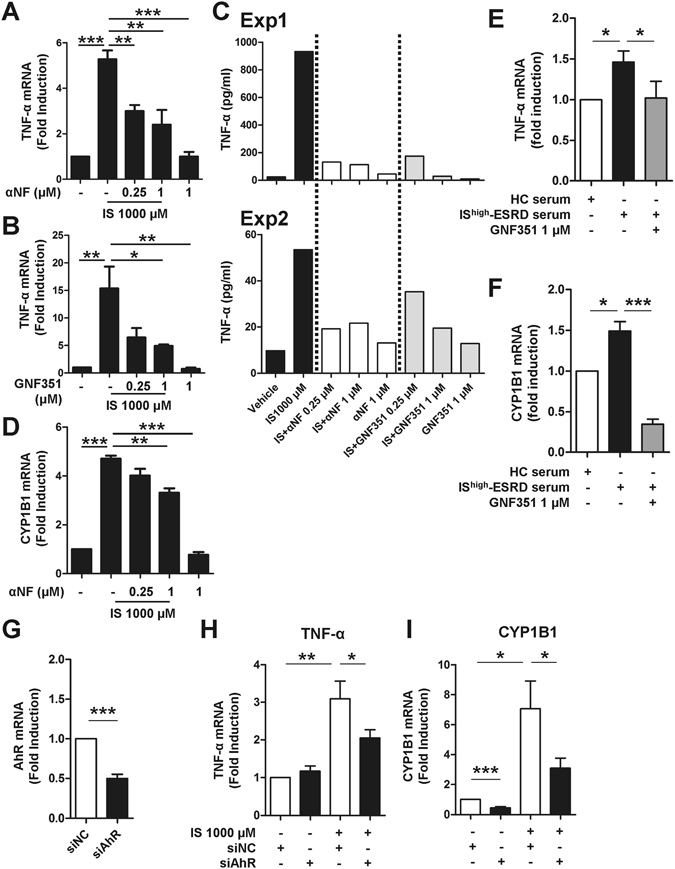
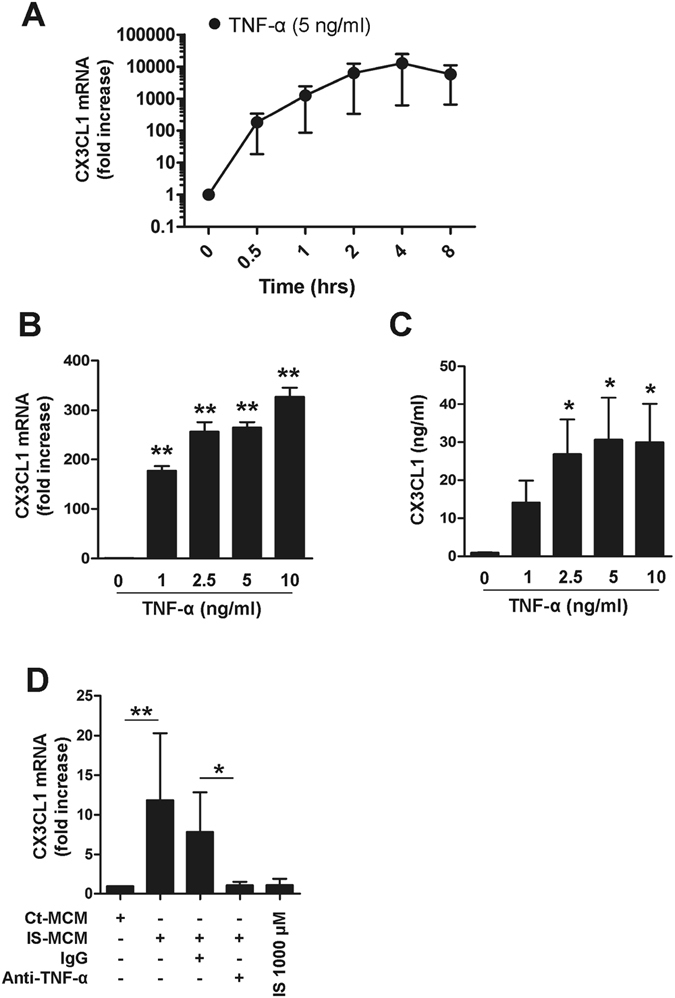
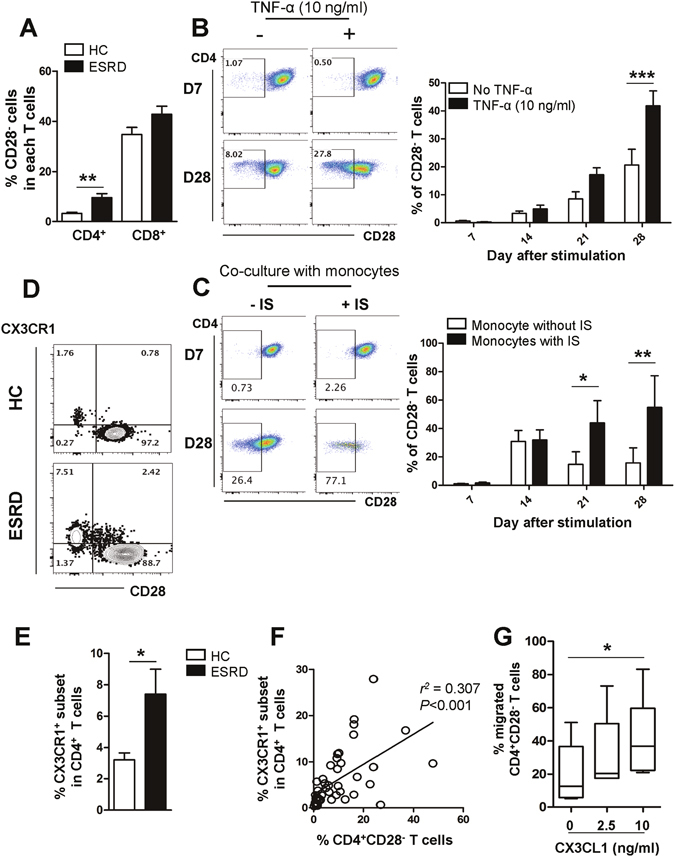
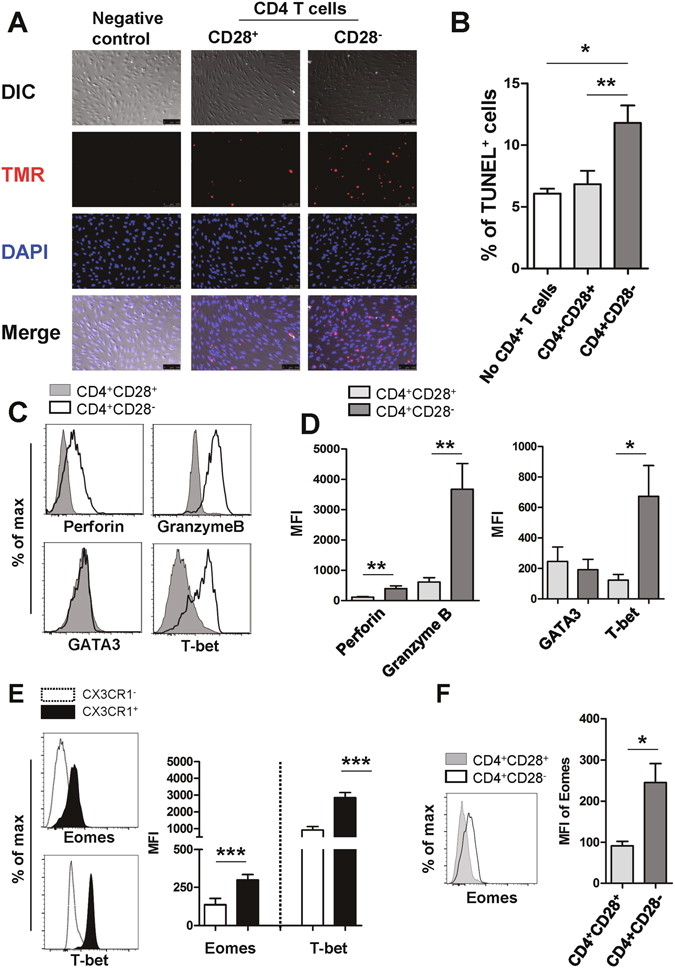
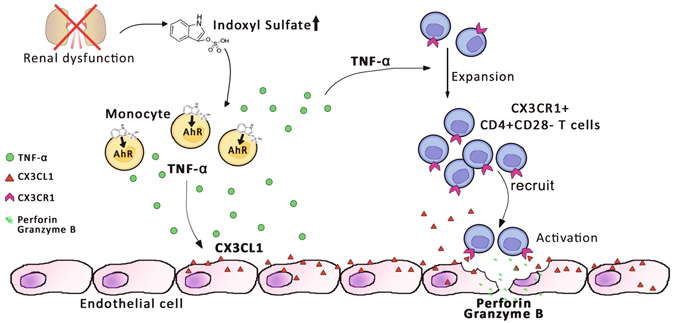
Progressive renal failure causes uremia-related immune dysfunction, which features a chronic inflammatory milieu. Given the central role of end-stage renal disease (ESRD)-related immune dysfunction in the pathogenesis of cardiovascular diseases (CVDs), much attention has been focused on how uremic toxins affect cellular immunity and the mechanisms underlying pathogenesis of atherosclerosis in ESRD patients. Here, we investigated the characteristics of monocytes and CD4+ T cells in ESRD patients and the immune responses induced by indoxyl sulfate (IS), a key uremic toxin, in order to explore the pathogenic effects of these cells on vascular endothelial cells. In ESRD patients, monocytes respond to IS through the aryl hydrocarbon receptor (AhR) and consequently produce increased levels of TNF-α. Upon stimulation with TNF-α, human vascular endothelial cells produce copious amounts of CX3CL1, a chemokine ligand of CX3CR1 that is highly expressed on CD4+CD28-T cells, the predominantly expanded cell type in ESRD patients. A migration assay showed that CD4+CD28- T cells were preferentially recruited by CX3CL1. Moreover, activated CD4+CD28- T cells exhibited cytotoxic capability allowing for the induction of apoptosis in HUVECs. Our findings suggest that in ESRD, IS-mediated immune dysfunction may cause vascular endothelial cell damage and thus, this toxin plays a pivotal role in the pathogenesis of CVD.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
-
- Meier P, Dayer E, Blanc E, Wauters JP. Early T cell activation correlates with expression of apoptosis markers in patients with end-stage renal disease. Journal of the American Society of Nephrology: JASN. 2002;13:204–212. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
