

Performances of Different Fragment Sizes for Reduced Representation Bisulfite Sequencing in Pigs
- PMID: 28596713
- PMCID: PMC5463379
- DOI: 10.1186/s12575-017-0054-5
Performances of Different Fragment Sizes for Reduced Representation Bisulfite Sequencing in Pigs
Abstract
Background: Reduced representation bisulfite sequencing (RRBS) has been widely used to profile genome-scale DNA methylation in mammalian genomes. However, the applications and technical performances of RRBS with different fragment sizes have not been systematically reported in pigs, which serve as one of the important biomedical models for humans. The aims of this study were to evaluate capacities of RRBS libraries with different fragment sizes to characterize the porcine genome.
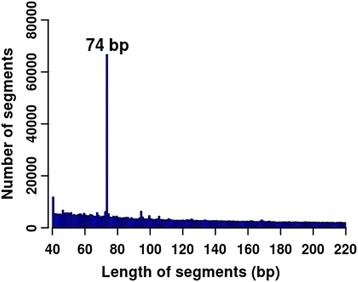
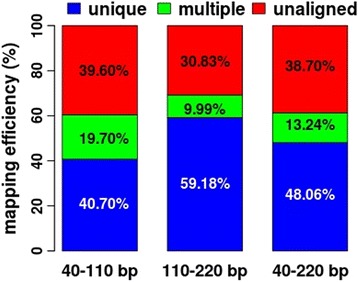
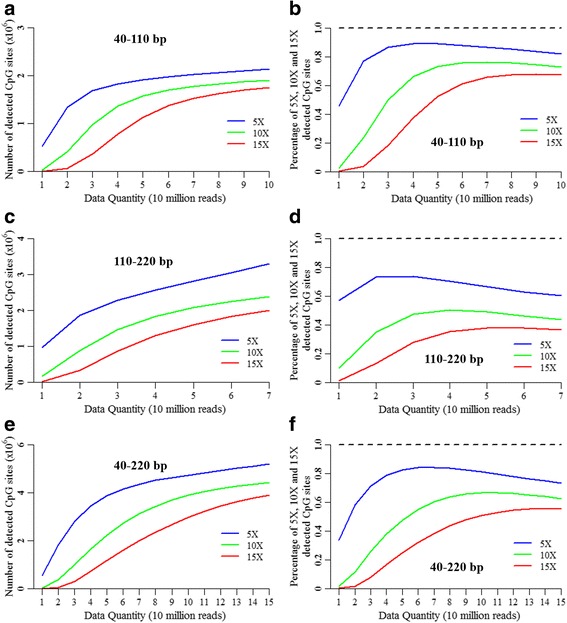
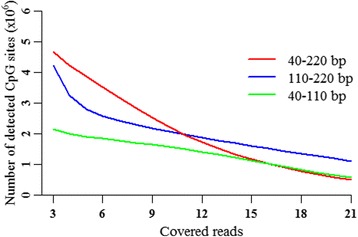
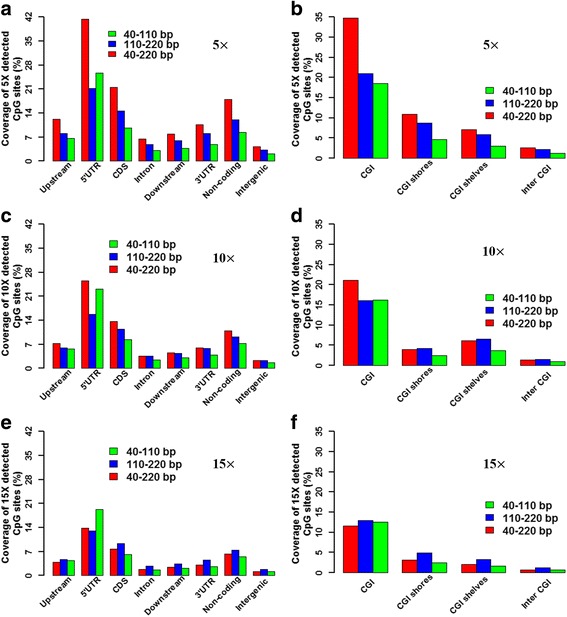
Results: We found that the MspI-digested segments between 40 and 220 bp harbored a high distribution peak at 74 bp, which were highly overlapped with the repetitive elements and might reduce the unique mapping alignment. The RRBS library of 110-220 bp fragment size had the highest unique mapping alignment and the lowest multiple alignment. The cost-effectiveness of the 40-110 bp, 110-220 bp and 40-220 bp fragment sizes might decrease when the dataset size was more than 70, 50 and 110 million reads for these three fragment sizes, respectively. Given a 50-million dataset size, the average sequencing depth of the detected CpG sites in the 110-220 bp fragment size appeared to be deeper than in the 40-110 bp and 40-220 bp fragment sizes, and these detected CpG sties differently located in gene- and CpG island-related regions.
Conclusions: In this study, our results demonstrated that selections of fragment sizes could affect the numbers and sequencing depth of detected CpG sites as well as the cost-efficiency. No single solution of RRBS is optimal in all circumstances for investigating genome-scale DNA methylation. This work provides the useful knowledge on designing and executing RRBS for investigating the genome-wide DNA methylation in tissues from pigs.
Keywords: DNA methylation; Different fragment sizes; Pigs; RRBS.
Figures
Similar articles
-
[Reduced representation bisulfite sequencing design for assessing the methylation of human CpG islands in large samples].Mol Biol (Mosk). 2015 Jul-Aug;49(4):689-99. doi: 10.7868/S0026898415040187. Mol Biol (Mosk). 2015. PMID: 26299869 Russian.
-
Mapping the zebrafish brain methylome using reduced representation bisulfite sequencing.Epigenetics. 2013 Sep;8(9):979-89. doi: 10.4161/epi.25797. Epub 2013 Jul 24. Epigenetics. 2013. PMID: 23975027 Free PMC article.
-
Preparation of reduced representation bisulfite sequencing libraries for genome-scale DNA methylation profiling.Nat Protoc. 2011 Apr;6(4):468-81. doi: 10.1038/nprot.2010.190. Epub 2011 Mar 18. Nat Protoc. 2011. PMID: 21412275
-
Adult porcine genome-wide DNA methylation patterns support pigs as a biomedical model.BMC Genomics. 2015 Oct 5;16:743. doi: 10.1186/s12864-015-1938-x. BMC Genomics. 2015. PMID: 26438392 Free PMC article.
-
A comprehensive evaluation of alignment software for reduced representation bisulfite sequencing data.Bioinformatics. 2018 Aug 15;34(16):2715-2723. doi: 10.1093/bioinformatics/bty174. Bioinformatics. 2018. PMID: 29579198
Cited by
-
Wild epigenetics: insights from epigenetic studies on natural populations.Proc Biol Sci. 2022 Feb 9;289(1968):20211633. doi: 10.1098/rspb.2021.1633. Epub 2022 Feb 9. Proc Biol Sci. 2022. PMID: 35135348 Free PMC article. Review.
-
Genome-Wide DNA Methylation Analysis of Hypothalamus During the Onset of Puberty in Gilts.Front Genet. 2019 Mar 19;10:228. doi: 10.3389/fgene.2019.00228. eCollection 2019. Front Genet. 2019. PMID: 30941164 Free PMC article.
-
DNA methylation patterns vary in boar sperm cells with different levels of DNA fragmentation.BMC Genomics. 2019 Nov 27;20(1):897. doi: 10.1186/s12864-019-6307-8. BMC Genomics. 2019. PMID: 31775629 Free PMC article.
-
Comparison between indicine and taurine cattle DNA methylation reveals epigenetic variation associated to differences in morphological adaptive traits.Epigenetics. 2023 Dec;18(1):2163363. doi: 10.1080/15592294.2022.2163363. Epub 2023 Jan 4. Epigenetics. 2023. PMID: 36600398 Free PMC article.
-
Genome-wide DNA methylation analysis of pituitaries during the initiation of puberty in gilts.PLoS One. 2019 Mar 7;14(3):e0212630. doi: 10.1371/journal.pone.0212630. eCollection 2019. PLoS One. 2019. PMID: 30845225 Free PMC article.
References
LinkOut - more resources
Full Text Sources
Other Literature Sources
