

Cullin 3SPOP ubiquitin E3 ligase promotes the poly-ubiquitination and degradation of HDAC6
- PMID: 28599312
- PMCID: PMC5564613
- DOI: 10.18632/oncotarget.18141
Cullin 3SPOP ubiquitin E3 ligase promotes the poly-ubiquitination and degradation of HDAC6
Abstract
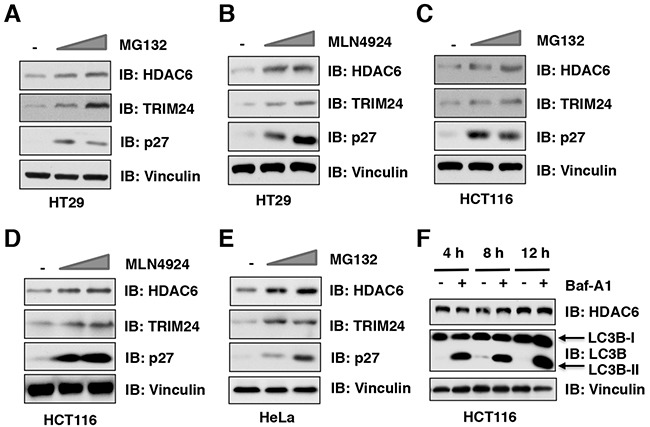
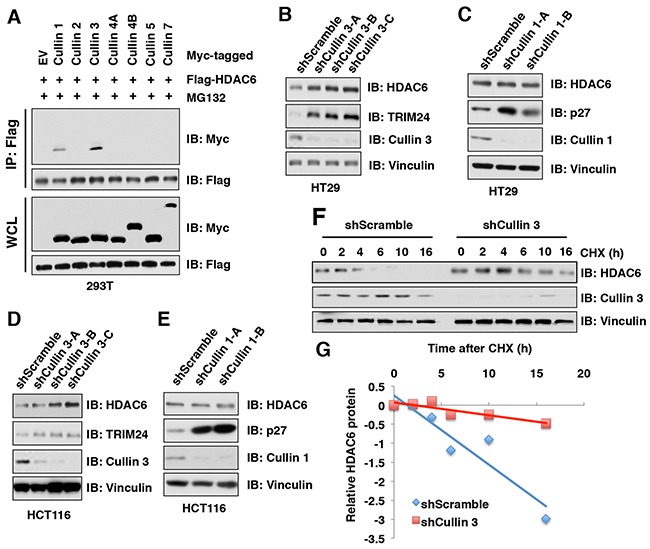
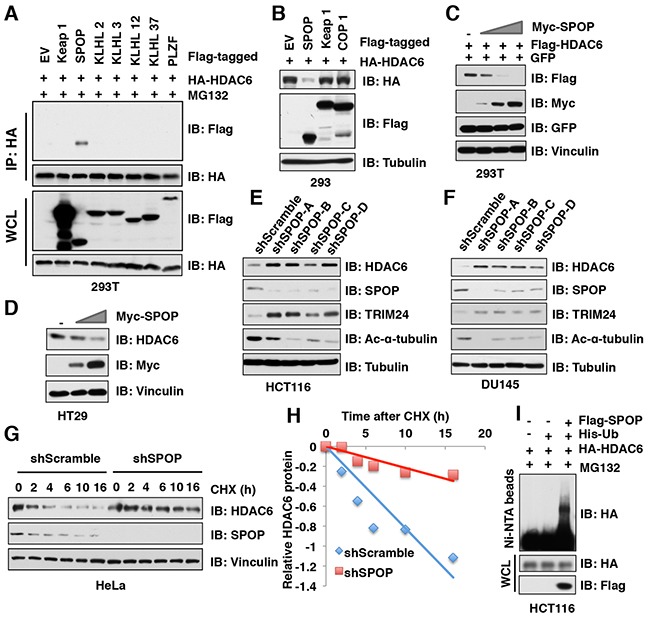
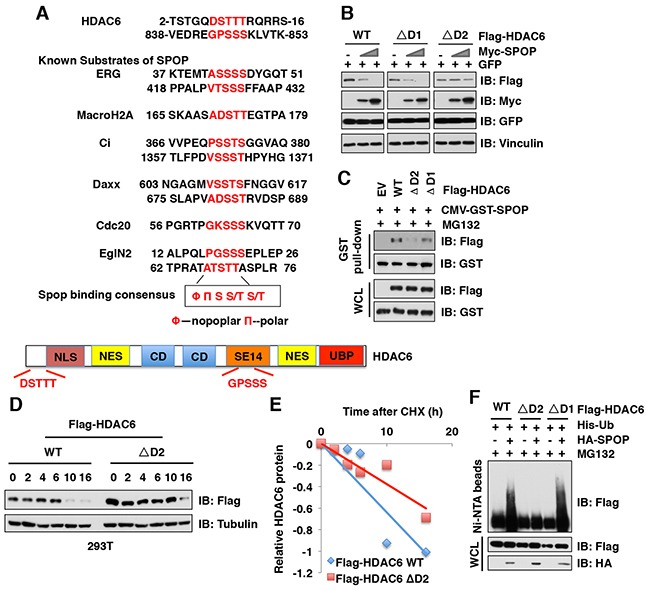
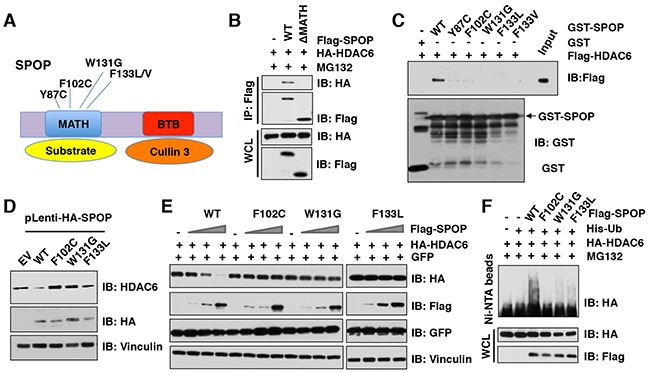
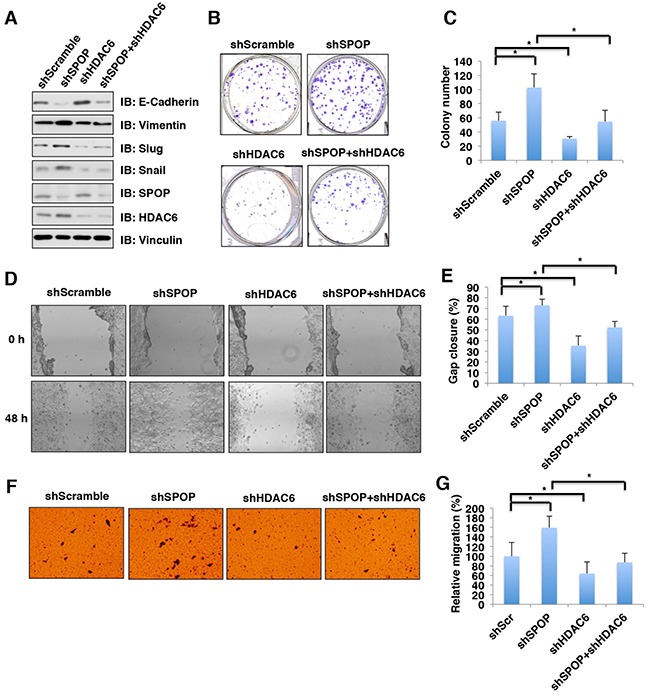
The histone deacetylase 6 (HDAC6) plays critical roles in human tumorigenesis and metastasis. As such, HDAC6-selective inhibitors have entered clinical trials for cancer therapy. However, the upstream regulator(s), especially ubiquitin E3 ligase(s), responsible for controlling the protein stability of HDAC6 remains largely undefined. Here, we report that Cullin 3SPOP earmarks HDAC6 for poly-ubiquitination and degradation. We found that the proteasome inhibitor MG132, or the Cullin-based E3 ligases inhibitor MLN4924, but not the autophagosome-lysosome inhibitor bafilomycin A1, stabilized endogenous HDAC6 protein in multiple cancer cell lines. Furthermore, we demonstrated that Cullin 3-based ubiquitin E3 ligase(s) primarily reduced the stability of HDAC6. Importantly, we identified SPOP, an adaptor protein of Cullin 3 family E3 ligases, specifically interacted with HDAC6, and promoted its poly-ubiquitination and subsequent degradation in cells. Notably, cancer-derived SPOP mutants disrupted their binding with HDAC6 and thereby failed to promote HDAC6 degradation. More importantly, increased cellular proliferation and migration in SPOP-depleted HCT116 colon cancer cells could be partly reversed by additional depletion of HDAC6, suggesting that HDAC6 is a key downstream effector for SPOP tumor suppressor function. Together, our data identify the tumor suppressor SPOP as an upstream negative regulator for HDAC6 stability, and SPOP loss-of-function mutations might lead to elevated levels of the HDAC6 oncoprotein to facilitate tumorigenesis and metastasis in various human cancers.
Keywords: Cullin 3; HDAC6; SPOP; tumorigenesis; ubiquitination.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
Similar articles
-
Functional analysis of Cullin 3 E3 ligases in tumorigenesis.Biochim Biophys Acta Rev Cancer. 2018 Jan;1869(1):11-28. doi: 10.1016/j.bbcan.2017.11.001. Epub 2017 Nov 8. Biochim Biophys Acta Rev Cancer. 2018. PMID: 29128526 Free PMC article. Review.
-
Prostate cancer-associated mutation in SPOP impairs its ability to target Cdc20 for poly-ubiquitination and degradation.Cancer Lett. 2017 Jan 28;385:207-214. doi: 10.1016/j.canlet.2016.10.021. Epub 2016 Oct 22. Cancer Lett. 2017. PMID: 27780719 Free PMC article.
-
Endometrial cancer-associated mutants of SPOP are defective in regulating estrogen receptor-α protein turnover.Cell Death Dis. 2015 Mar 12;6(3):e1687. doi: 10.1038/cddis.2015.47. Cell Death Dis. 2015. PMID: 25766326 Free PMC article.
-
Breast cancer metastasis suppressor 1 (BRMS1) is destabilized by the Cul3-SPOP E3 ubiquitin ligase complex.Biochem Biophys Res Commun. 2011 Dec 2;415(4):720-6. doi: 10.1016/j.bbrc.2011.10.154. Epub 2011 Nov 9. Biochem Biophys Res Commun. 2011. PMID: 22085717
-
Cullin-RING E3 Ubiquitin Ligases: Bridges to Destruction.Subcell Biochem. 2017;83:323-347. doi: 10.1007/978-3-319-46503-6_12. Subcell Biochem. 2017. PMID: 28271482 Free PMC article. Review.
Cited by
-
Role of HDAC6 and Its Selective Inhibitors in Gastrointestinal Cancer.Front Cell Dev Biol. 2021 Dec 2;9:719390. doi: 10.3389/fcell.2021.719390. eCollection 2021. Front Cell Dev Biol. 2021. PMID: 34938729 Free PMC article. Review.
-
SPOP and cancer: a systematic review.Am J Cancer Res. 2020 Mar 1;10(3):704-726. eCollection 2020. Am J Cancer Res. 2020. PMID: 32266086 Free PMC article. Review.
-
Functional analysis of Cullin 3 E3 ligases in tumorigenesis.Biochim Biophys Acta Rev Cancer. 2018 Jan;1869(1):11-28. doi: 10.1016/j.bbcan.2017.11.001. Epub 2017 Nov 8. Biochim Biophys Acta Rev Cancer. 2018. PMID: 29128526 Free PMC article. Review.
-
Pin1 coordinates HDAC6 upregulation with cell migration in lung cancer cells.Int J Med Sci. 2020 Sep 21;17(17):2635-2643. doi: 10.7150/ijms.50097. eCollection 2020. Int J Med Sci. 2020. PMID: 33162791 Free PMC article.
-
SPOP in Cancer: Phenomena, Mechanisms and Its Role in Therapeutic Implications.Genes (Basel). 2022 Nov 7;13(11):2051. doi: 10.3390/genes13112051. Genes (Basel). 2022. PMID: 36360288 Free PMC article. Review.
References
-
- Glozak MA, Seto E. Histone deacetylases and cancer. Oncogene. 2007;26:5420–5432. - PubMed
-
- Ganai SA. Designing isoform-selective inhibitors against Classical HDACs for effective anticancer therapy: Insight and perspectives from in silico. Curr Drug Targets. 2017 Jan 12; [Epub ahead of print] - PubMed
-
- Batchu SN, Brijmohan AS, Advani A. The therapeutic hope for HDAC6 inhibitors in malignancy and chronic disease. Clin Sci (Lond) 2016;130:987–1003. - PubMed
-
- Seidel C, Schnekenburger M, Dicato M, Diederich M. Histone deacetylase 6 in health and disease. Epigenomics. 2015;7:103–118. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
