

An Optimized Shotgun Strategy for the Rapid Generation of Comprehensive Human Proteomes
- PMID: 28601559
- PMCID: PMC5493283
- DOI: 10.1016/j.cels.2017.05.009
An Optimized Shotgun Strategy for the Rapid Generation of Comprehensive Human Proteomes
Abstract
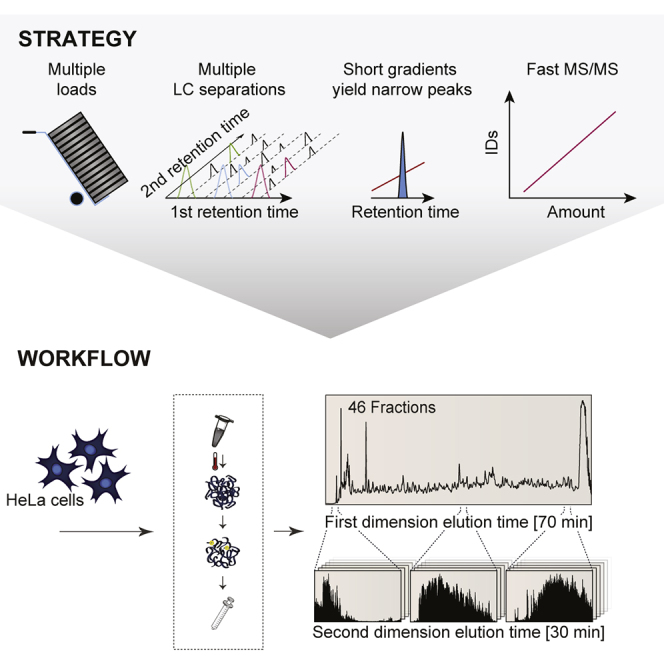
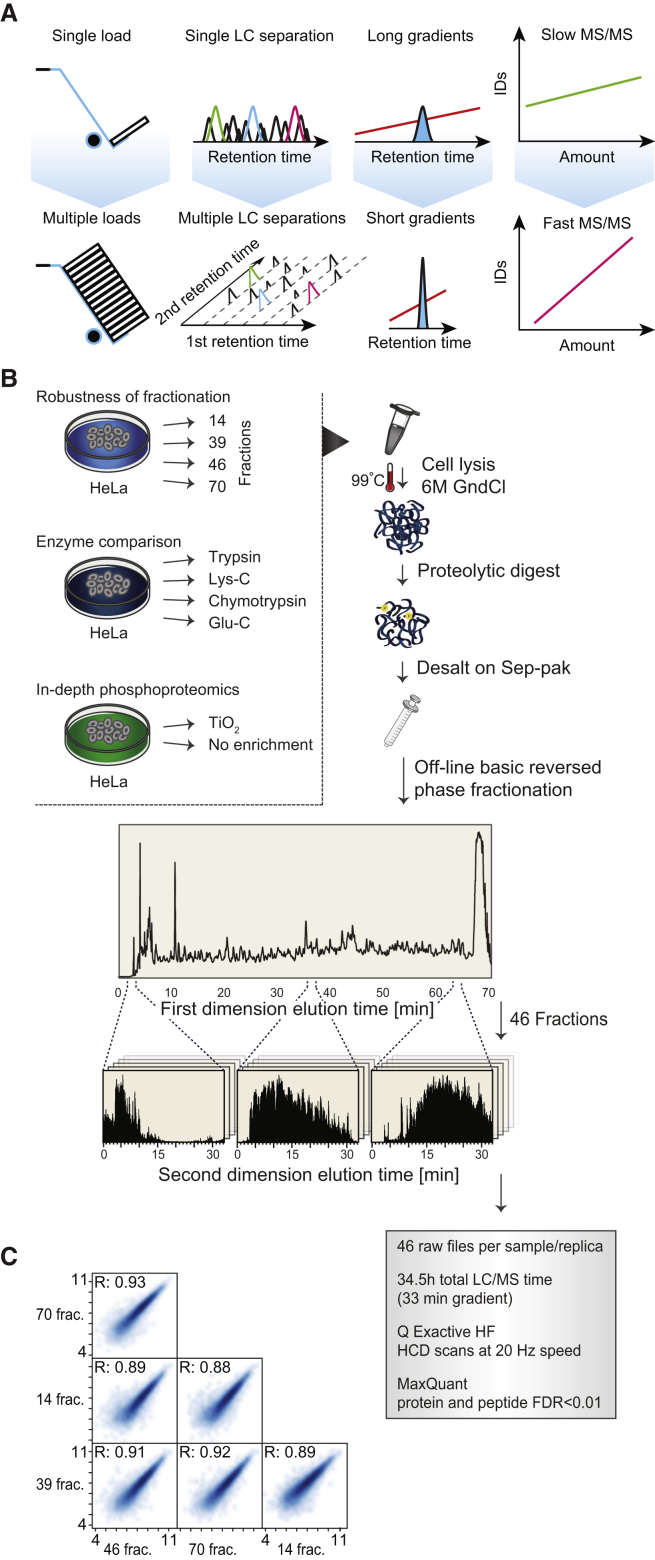
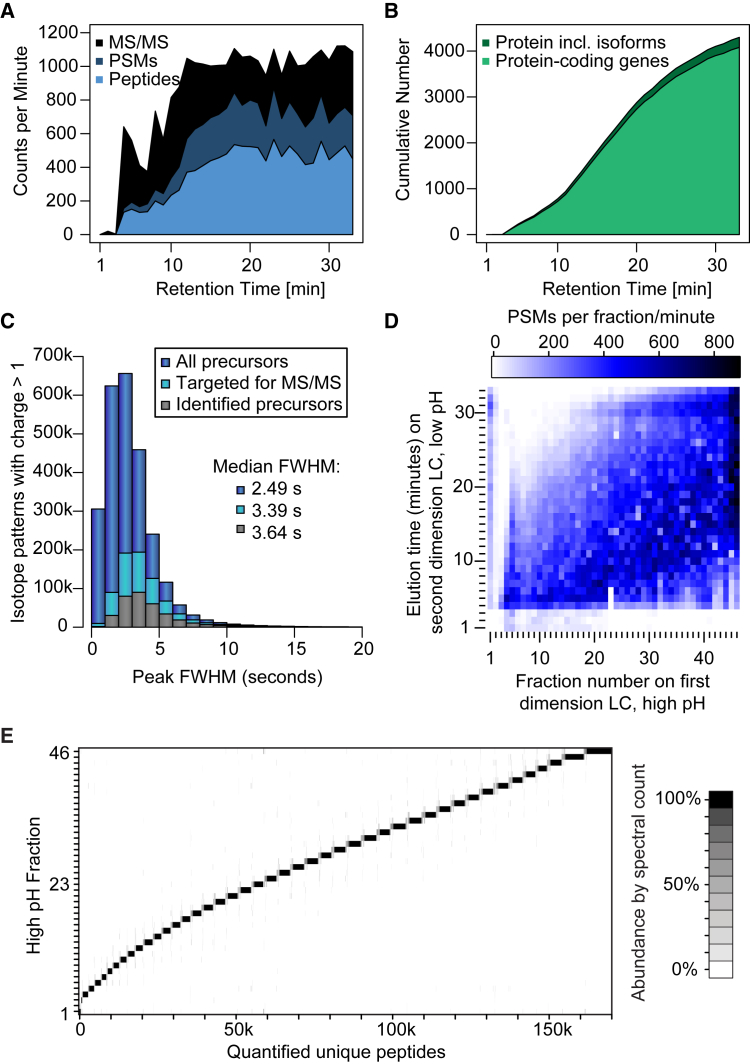
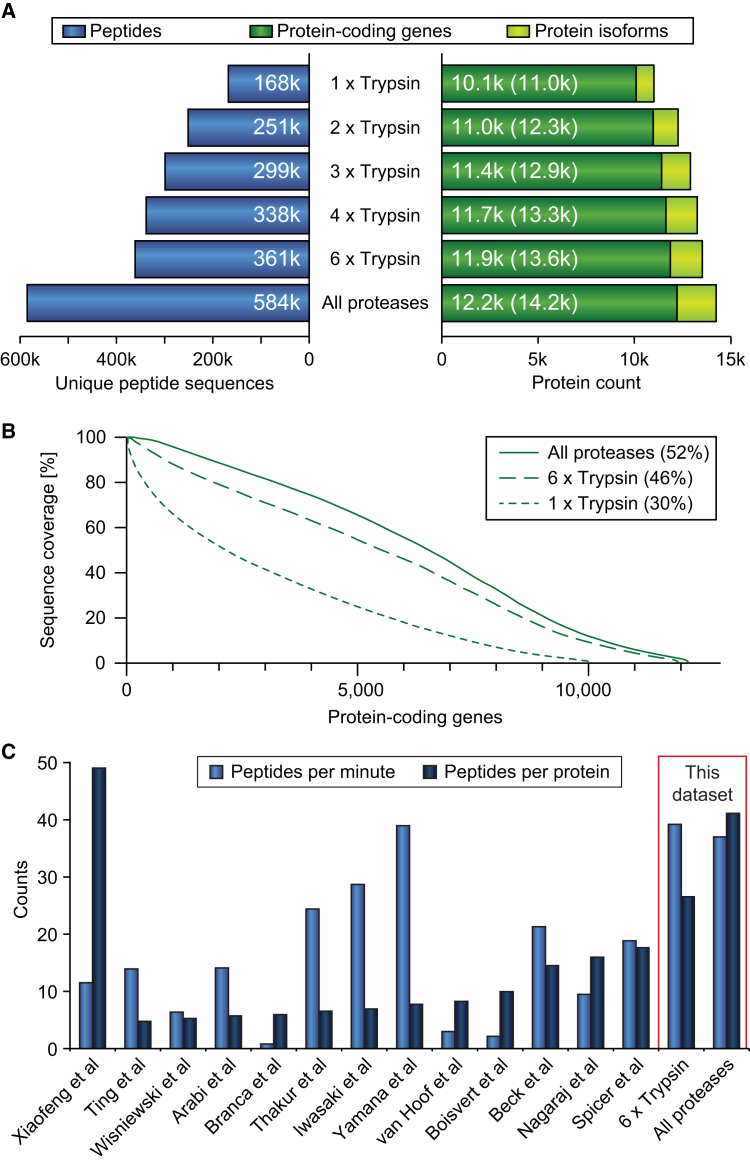
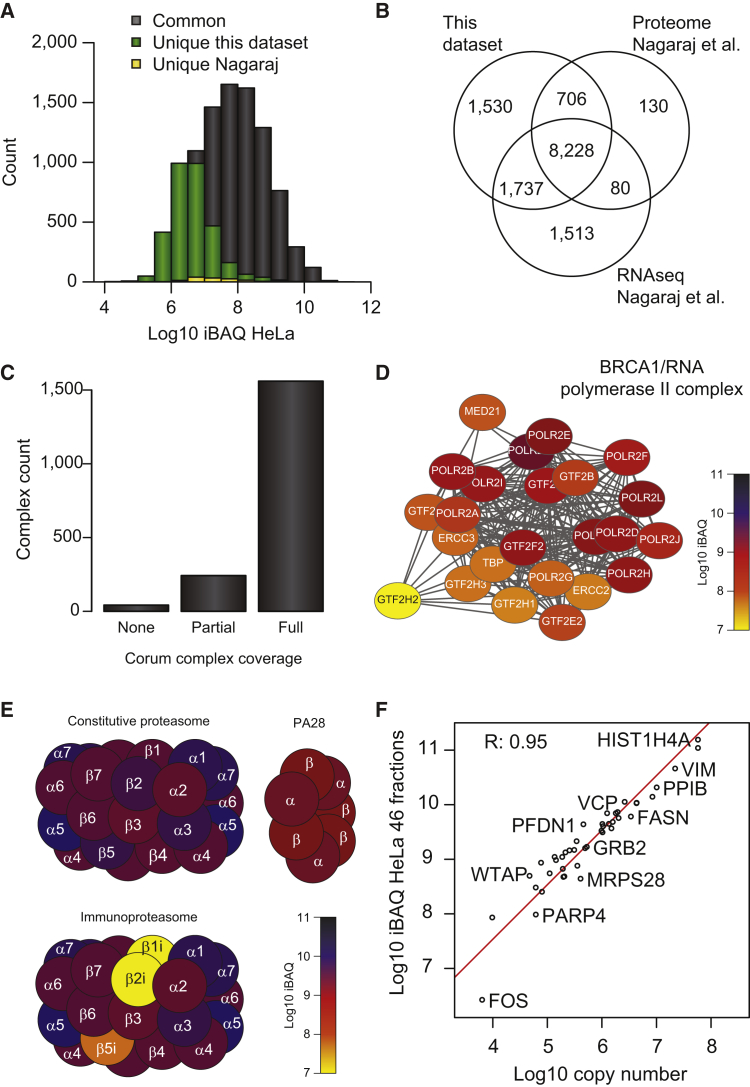
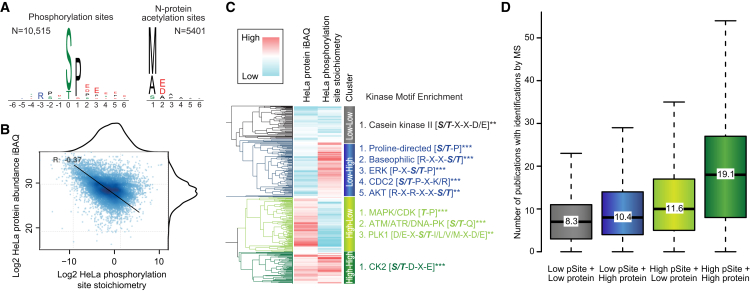
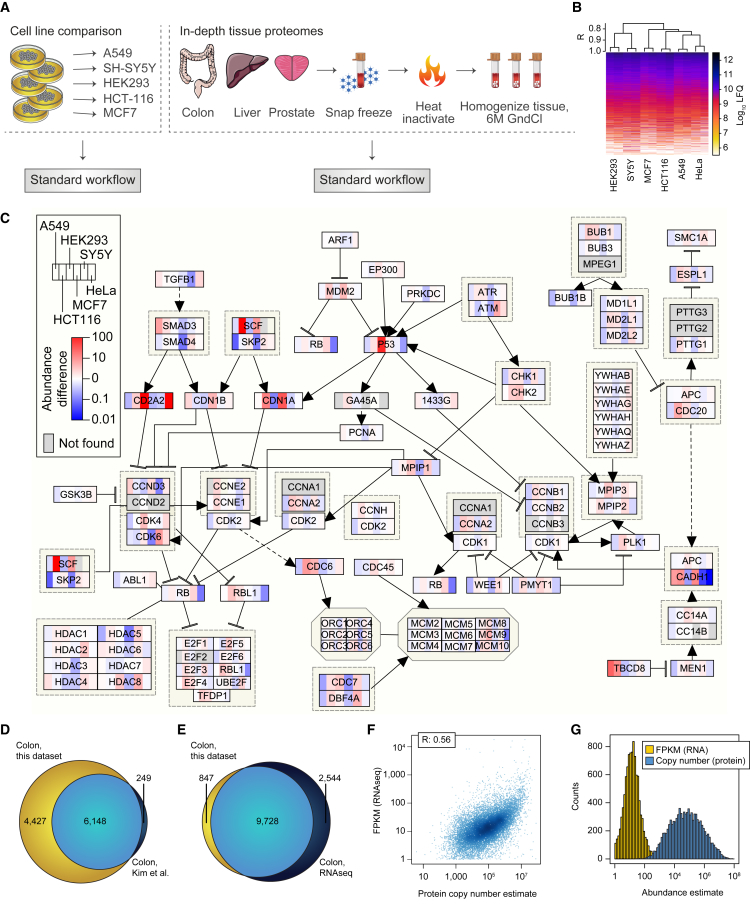
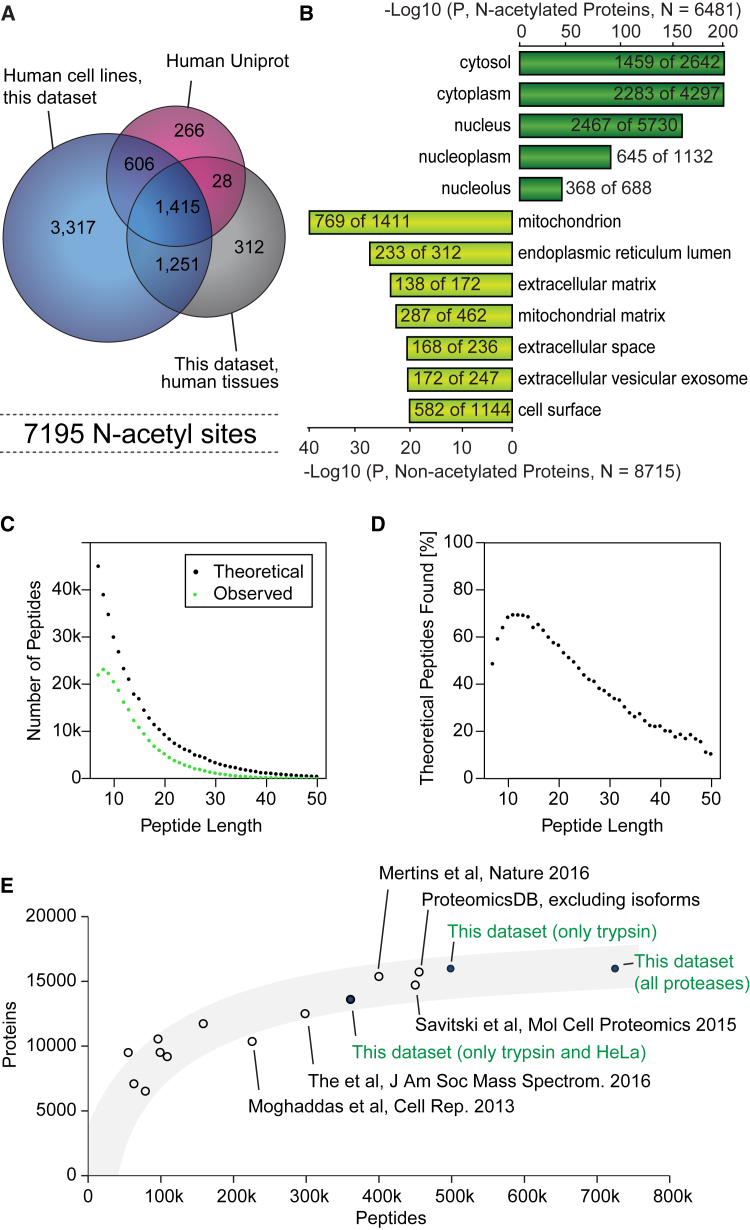
This study investigates the challenge of comprehensively cataloging the complete human proteome from a single-cell type using mass spectrometry (MS)-based shotgun proteomics. We modify a classical two-dimensional high-resolution reversed-phase peptide fractionation scheme and optimize a protocol that provides sufficient peak capacity to saturate the sequencing speed of modern MS instruments. This strategy enables the deepest proteome of a human single-cell type to date, with the HeLa proteome sequenced to a depth of ∼584,000 unique peptide sequences and ∼14,200 protein isoforms (∼12,200 protein-coding genes). This depth is comparable with next-generation RNA sequencing and enables the identification of post-translational modifications, including ∼7,000 N-acetylation sites and ∼10,000 phosphorylation sites, without the need for enrichment. We further demonstrate the general applicability and clinical potential of this proteomics strategy by comprehensively quantifying global proteome expression in several different human cancer cell lines and patient tissue samples.
Keywords: HeLa; PTM; acetylation; high pH reversed-phase fractionation; human proteome; mass spectrometry; orbitrap; patient samples; phosphorylation; proteomics.
Copyright © 2017 The Author(s). Published by Elsevier Inc. All rights reserved.
Figures
Comment in
-
New Apex in Proteome Analysis.Cell Syst. 2017 Jun 28;4(6):581-582. doi: 10.1016/j.cels.2017.06.009. Cell Syst. 2017. PMID: 28662382
References
-
- Batth T.S., Francavilla C., Olsen J.V. Off-line high-pH reversed-phase fractionation for in-depth phosphoproteomics. J. Proteome Res. 2014;13:6176–6186. - PubMed
-
- Branca R.M.M., Orre L.M., Johansson H.J., Granholm V., Huss M., Perez-Bercoff A., Forshed J., Kall L., Lehtio J. HiRIEF LC-MSMS enables deep proteome coverage and unbiased proteogenomics. Nat. Methods. 2014;11:59–62. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
