

Ultra-Early Phase pathologies of Alzheimer's disease and other neurodegenerative diseases
- PMID: 28603208
- PMCID: PMC5709537
- DOI: 10.2183/pjab.93.022
Ultra-Early Phase pathologies of Alzheimer's disease and other neurodegenerative diseases
Abstract
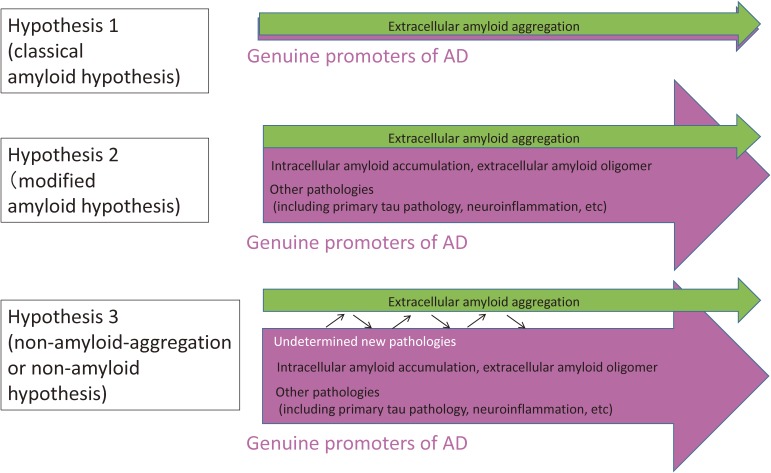
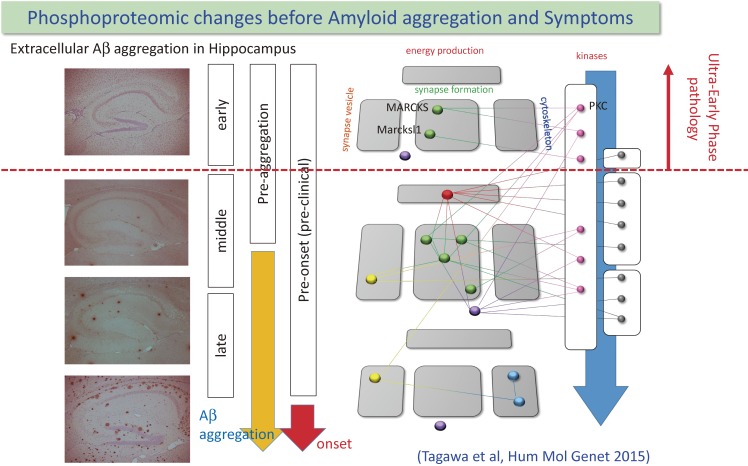
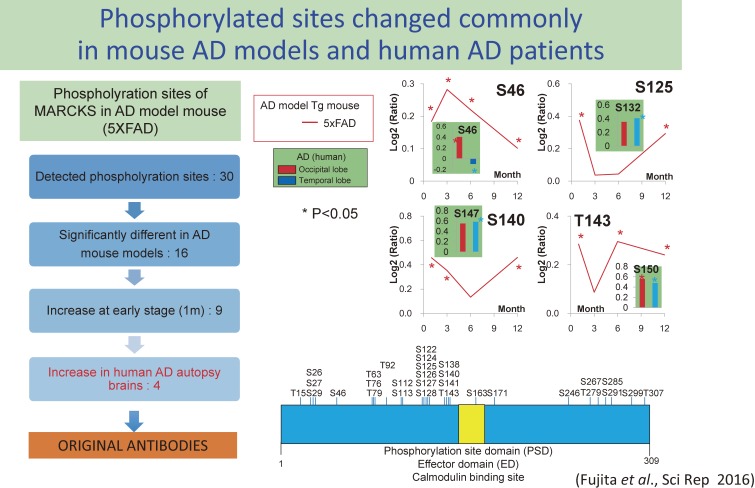
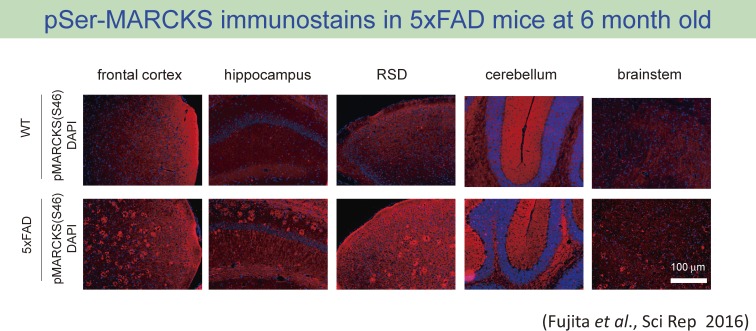
The concept of neurodegenerative diseases and the therapeutics targeting these intractable diseases are changing rapidly. Protein aggregation as the top of pathological cascade is now challenged, and many alternative ideas are proposed. Early molecular pathologies before microscopic detection of diseases protein aggregates, which I propose to call "Ultra-Early Phase pathologies or phase 0 pathologies", are the focus of research that might explain the failures of clinical trials with anti-Aβ antibodies against Alzheimer's disease. In this review article, I summarize the critical issues that should be successfully and consistently answered by a new concept of neurodegeneration. For reevaluating old concepts and reconstructing a new concept of neurodegeneration that will replace the old ones, non-biased comprehensive approaches including proteome combined with systems biology analyses will be a powerful tool. I introduce our recent efforts in this orientation that have reached to the stage of non-clinical proof of concept applicable to clinical trials.
Keywords: Alzheimer’s disease; Huntington’s disease; Ultra-Early Phase pathology; amyloid hypothesis; intracellular amyloid; neurodegeneration.
Figures
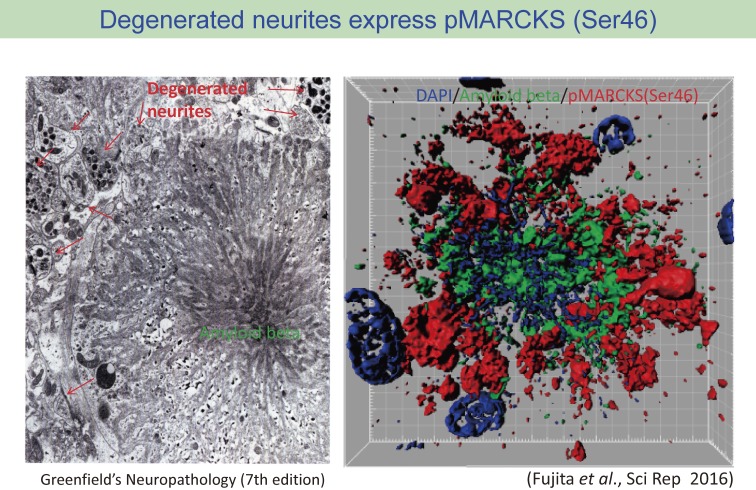
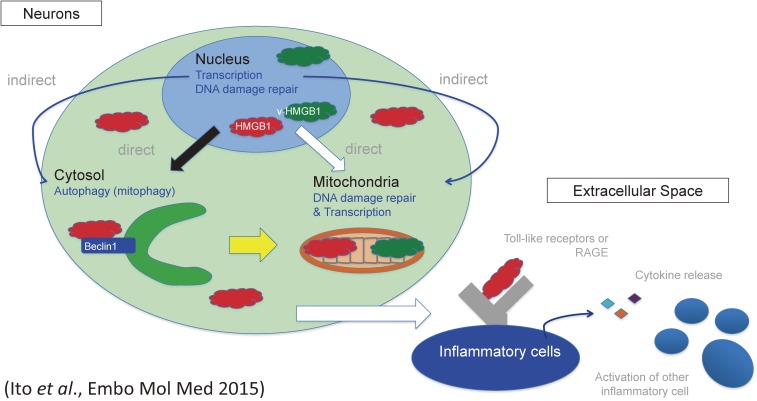
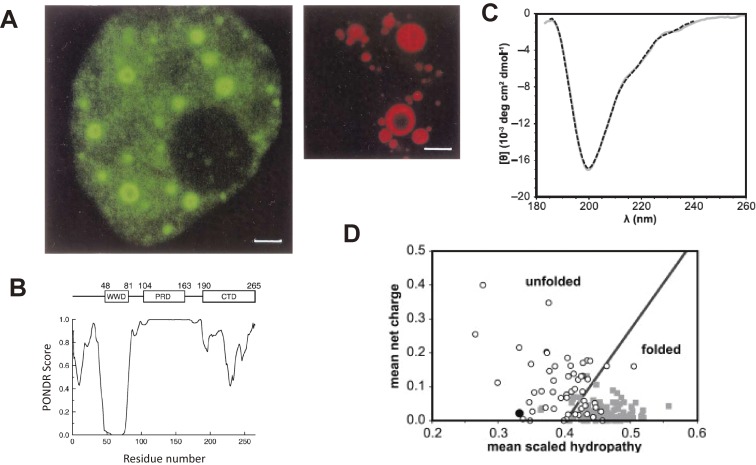
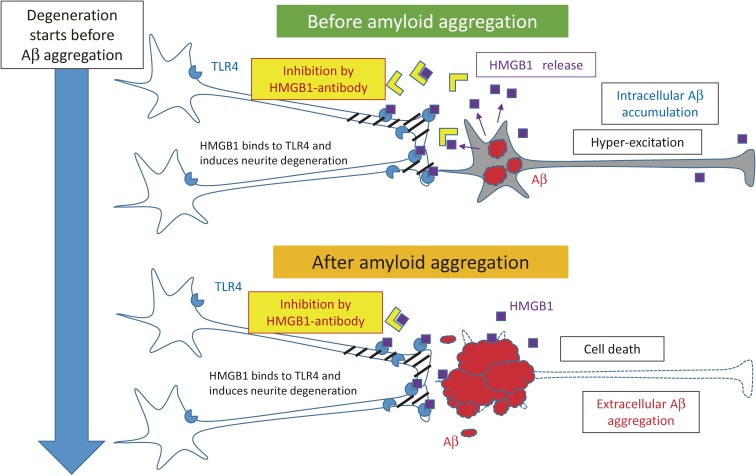
References
-
- Halliday G., Flowers D., Baum L. (1994) Analysis of staining methods for different cortical plaques in Alzheimer’s disease. Acta Neuropathol. 87, 174–186. - PubMed
-
- Ponte P., Gonzalez-DeWhitt P., Schilling J., Miller J., Hsu D., Greenberg B., Davis K., Wallace W., Lieberburg I., Fuller F. (1988) A new A4 amyloid mRNA contains a domain homologous to serine proteinase inhibitors. Nature 331, 525–527. - PubMed
-
- Kitaguchi N., Takahashi Y., Tokushima Y., Shiojiri S., Ito H. (1988) Novel precursor of Alzheimer’s disease amyloid protein shows protease inhibitory activity. Nature 331, 530–532. - PubMed
-
- Manning R.W., Reid C.M., Lampe R.A., Davis L.G. (1988) Identification in rodents and other species of an mRNA homologous to the human beta-amyloid precursor. Brain Res. 427, 293–297. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous