

Galactosialidosis: historic aspects and overview of investigated and emerging treatment options
- PMID: 28603679
- PMCID: PMC5461780
- DOI: 10.1080/21678707.2016.1266933
Galactosialidosis: historic aspects and overview of investigated and emerging treatment options
Abstract
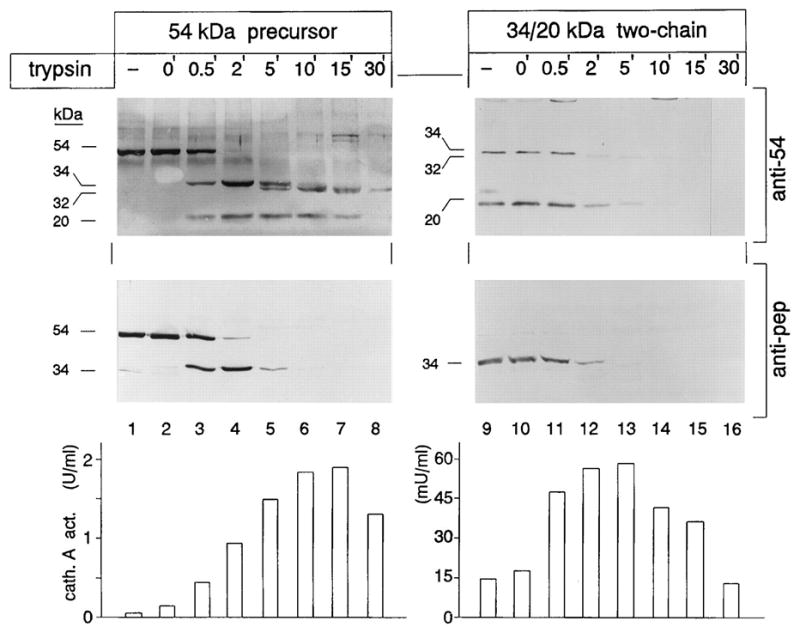
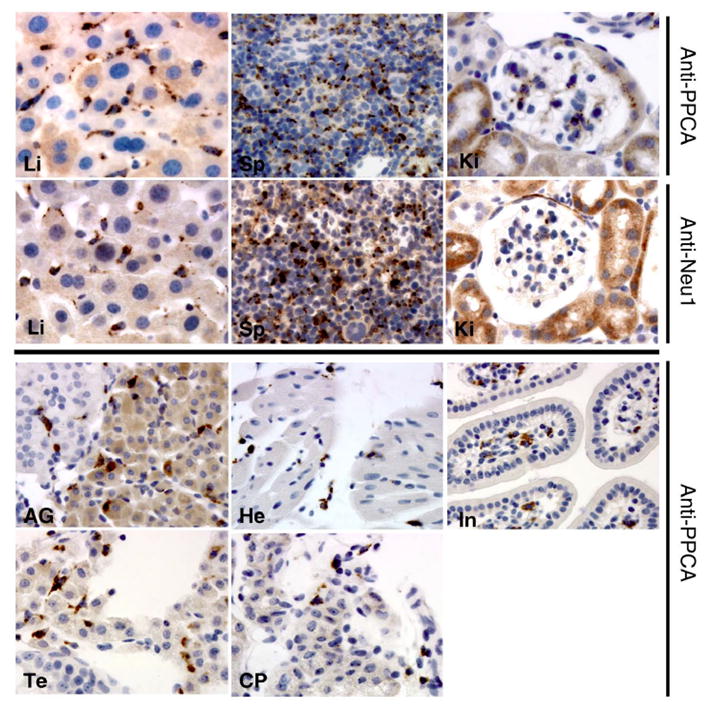
Introduction: Galactosialidosis is a glycoprotein storage disease caused by mutations in the CTSA gene, encoding lysosomal protective protein/cathepsin A (PPCA). The enzyme's catalytic activity is distinct from its protective function towards β-galactosidase (β-GAL) and neuraminidase 1 (NEU1), with which PPCA forms a complex. In this configuration the two glycosidases acquire their full activity and stability in lysosomes. Deficiency of PPCA results in combined NEU1/β-GAL deficiency. Because of its low incidence, galactosialidosis is considered an orphan disorder with no therapy yet available.
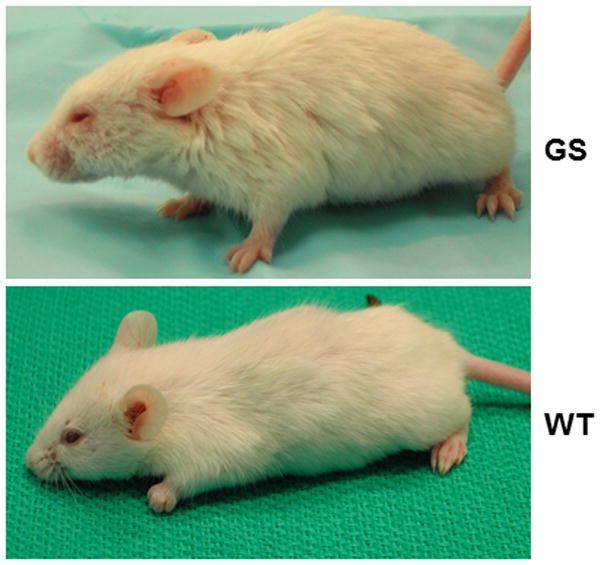
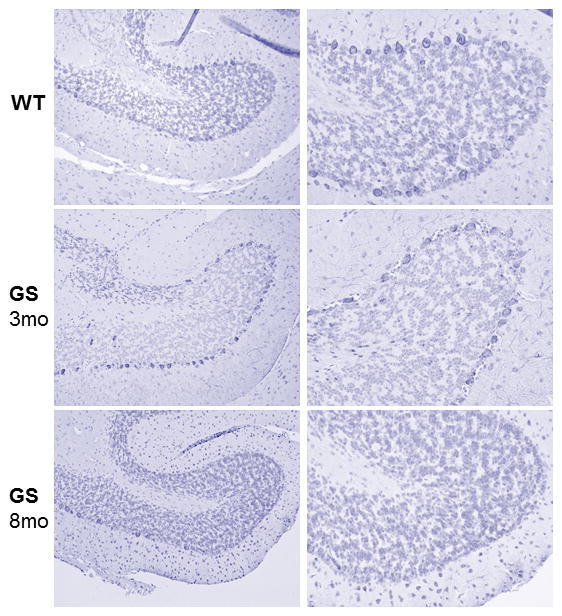
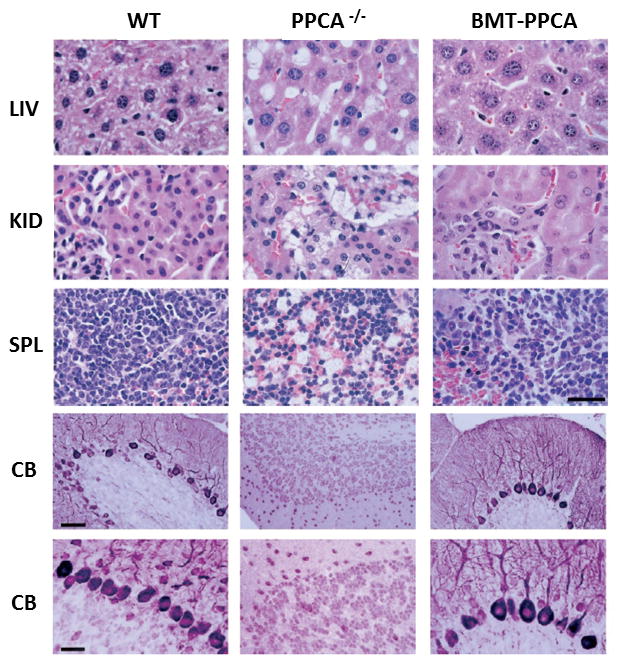
Areas covered: This review gives a historic overview on the discovery of PPCA, which defined galactosialidosis as a new clinical entity; the evidence for the existence of the PPCA/NEU1/β-GAL complex; the clinical forms of galactosialidosis and disease-causing CTSA mutations. Ppca-/- mice have proven to be a suitable model to test different therapeutic approaches, paving the way for the development of clinical trials for patients with galactosialidosis.
Expert opinion: Improved understanding of the molecular bases of disease has sparked renewed incentive from clinicians and scientists alike to develop therapies for rare conditions, like GS, and has increased the willingness of biotech companies to invest in the manufacturing of new therapeutics. Both ERT and gene therapy may become available to patients in the near future.
Keywords: CTSA; PPCA; galactosialidosis; lysosomal storage disease; therapy.
Conflict of interest statement
Declaration of interest A.d’A. holds the Jewelers for Children Endowed Chair in Genetics and Gene Therapy. The authors have no other relevant affiliations or financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Figures
References
-
- Derry DM, Fawcett JS, Andermann F, et al. Late infantile systemic lipidosis. Major monosialogangliosidosis. Delineation of two types. Neurology. 1968 Apr;18(4):340–348. - PubMed
-
- Wenger DA, Goodman SI, Myers GG. Letter: beta-galactosidase deficiency in young adults. Lancet. 1974 Nov 30;2(7892):1319–1320. - PubMed
-
- Wenger DA, Tarby TJ, Wharton C. Macular cherry-red spots and myoclonus with dementia: coexistent neuraminidase and beta galactosidase deficiencies. Biochem Biophys Res Commun. 1978 May 30;82(2):589–595. - PubMed
-
- Hoogeveen A, d’Azzo A, Brossmer R, et al. Correction of combined beta-galactosidase/neuraminidase deficiency in human fibroblasts. Biochem Biophys Res Commun. 1981 Nov 16;103(1):292–300. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous