

Genetic diagnosis of Mendelian disorders via RNA sequencing
- PMID: 28604674
- PMCID: PMC5499207
- DOI: 10.1038/ncomms15824
Genetic diagnosis of Mendelian disorders via RNA sequencing
Abstract
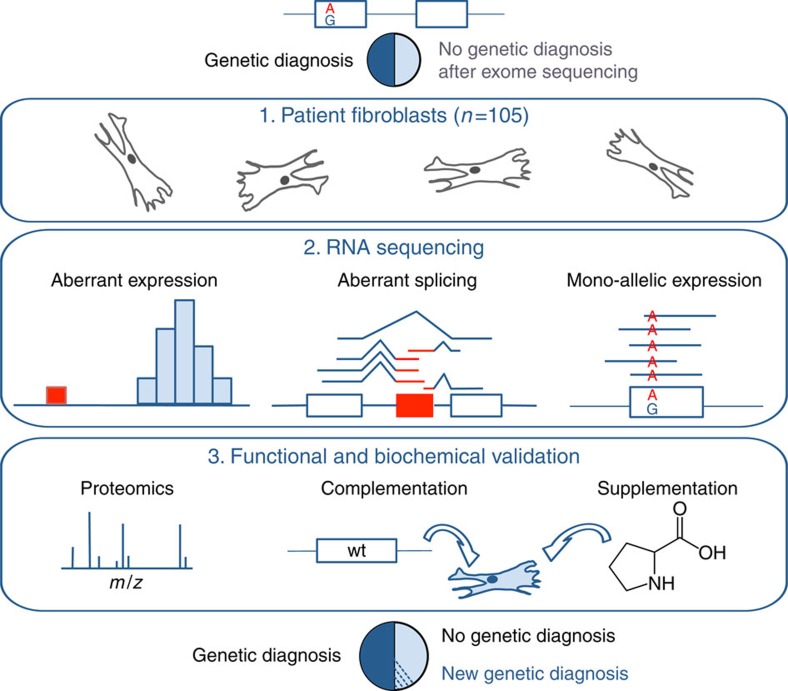
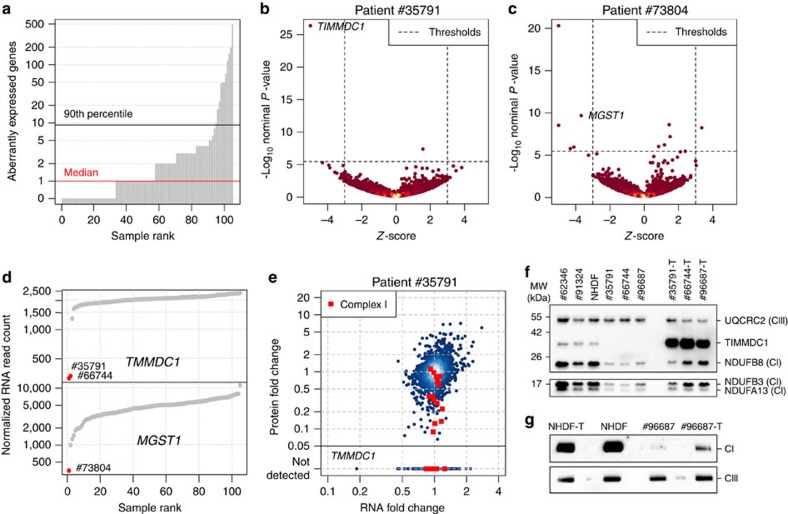
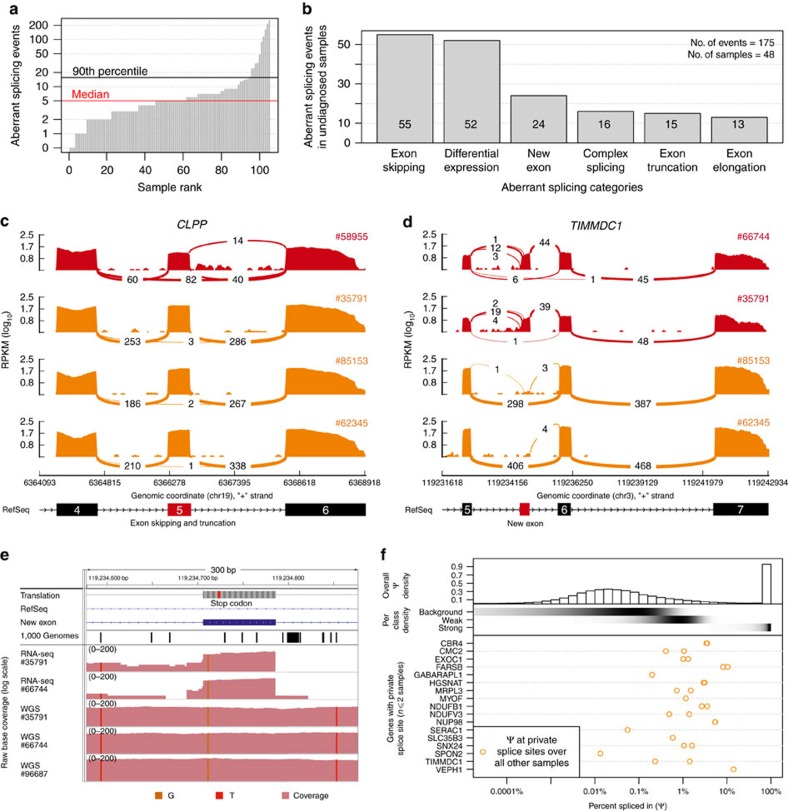
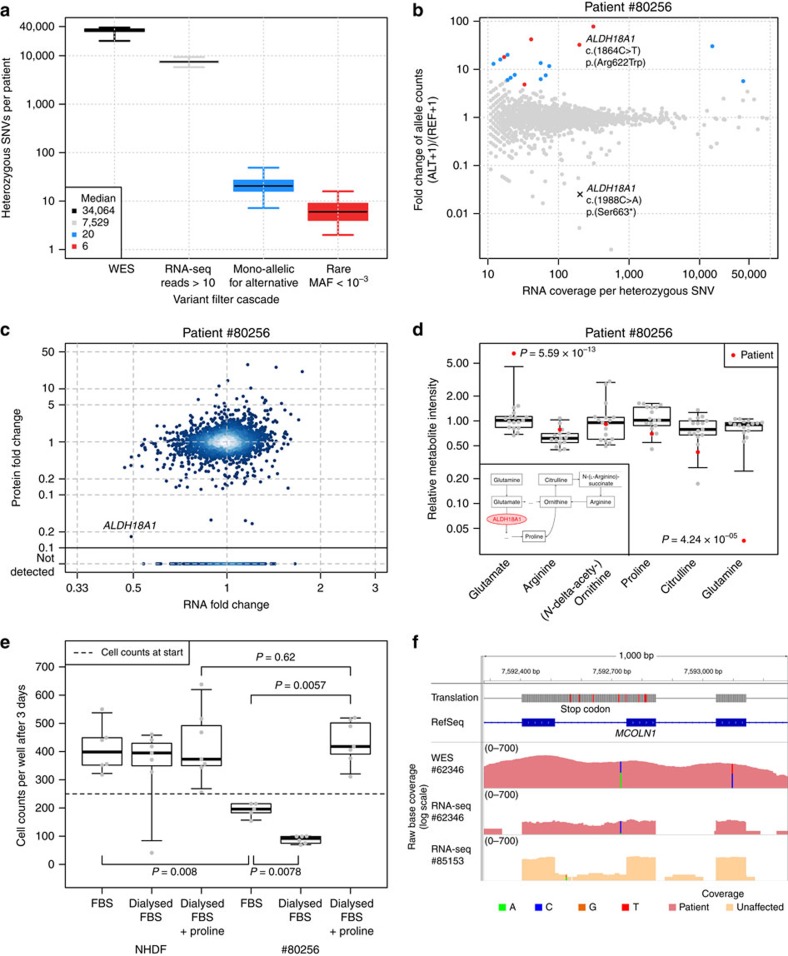
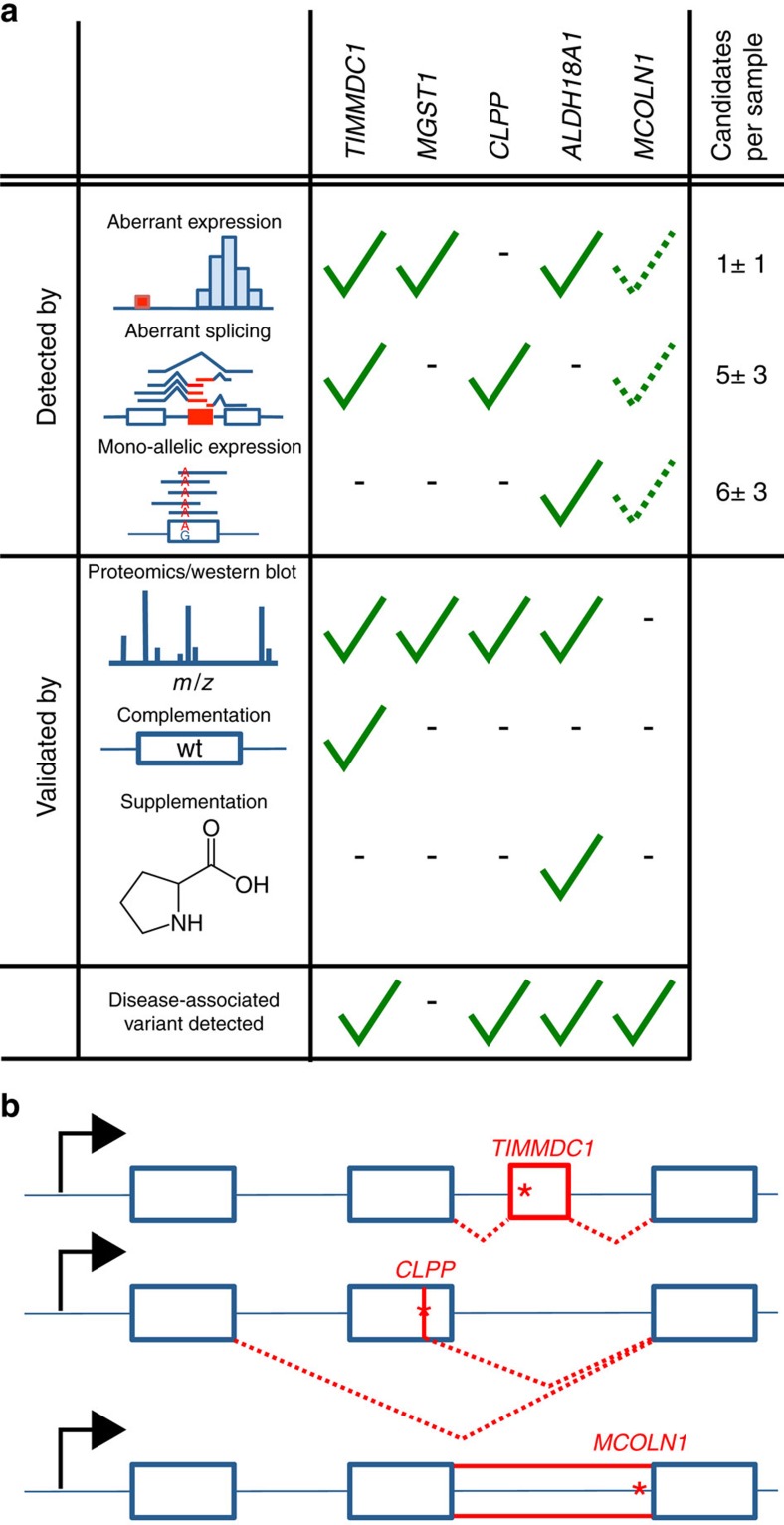
Across a variety of Mendelian disorders, ∼50-75% of patients do not receive a genetic diagnosis by exome sequencing indicating disease-causing variants in non-coding regions. Although genome sequencing in principle reveals all genetic variants, their sizeable number and poorer annotation make prioritization challenging. Here, we demonstrate the power of transcriptome sequencing to molecularly diagnose 10% (5 of 48) of mitochondriopathy patients and identify candidate genes for the remainder. We find a median of one aberrantly expressed gene, five aberrant splicing events and six mono-allelically expressed rare variants in patient-derived fibroblasts and establish disease-causing roles for each kind. Private exons often arise from cryptic splice sites providing an important clue for variant prioritization. One such event is found in the complex I assembly factor TIMMDC1 establishing a novel disease-associated gene. In conclusion, our study expands the diagnostic tools for detecting non-exonic variants and provides examples of intronic loss-of-function variants with pathological relevance.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
