

A TNF-JNK-Axl-ERK signaling axis mediates primary resistance to EGFR inhibition in glioblastoma
- PMID: 28604685
- PMCID: PMC5529219
- DOI: 10.1038/nn.4584
A TNF-JNK-Axl-ERK signaling axis mediates primary resistance to EGFR inhibition in glioblastoma
Abstract
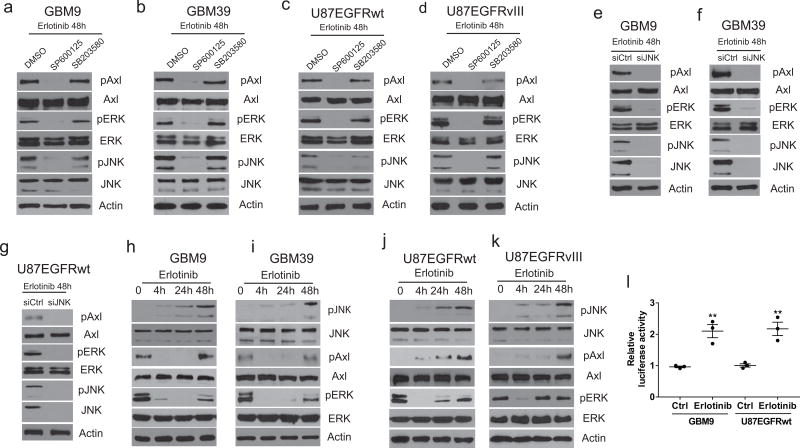
Aberrant epidermal growth factor receptor (EGFR) signaling is widespread in cancer, making the EGFR an important target for therapy. EGFR gene amplification and mutation are common in glioblastoma (GBM), but EGFR inhibition has not been effective in treating this tumor. Here we propose that primary resistance to EGFR inhibition in glioma cells results from a rapid compensatory response to EGFR inhibition that mediates cell survival. We show that in glioma cells expressing either EGFR wild type or the mutant EGFRvIII, EGFR inhibition triggers a rapid adaptive response driven by increased tumor necrosis factor (TNF) secretion, which leads to activation in turn of c-Jun N-terminal kinase (JNK), the Axl receptor tyrosine kinase and extracellular signal-regulated kinases (ERK). Inhibition of this adaptive axis at multiple nodes rendered glioma cells with primary resistance sensitive to EGFR inhibition. Our findings provide a possible explanation for the failures of anti-EGFR therapy in GBM and suggest a new approach to the treatment of EGFR-expressing GBM using a combination of EGFR and TNF inhibition.
Figures






Comment in
-
Helping EGFR inhibition to block cancer.Nat Neurosci. 2017 Jul 26;20(8):1035-1037. doi: 10.1038/nn.4605. Nat Neurosci. 2017. PMID: 28745721 No abstract available.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
