

EMT cells increase breast cancer metastasis via paracrine GLI activation in neighbouring tumour cells
- PMID: 28604738
- PMCID: PMC5472791
- DOI: 10.1038/ncomms15773
EMT cells increase breast cancer metastasis via paracrine GLI activation in neighbouring tumour cells
Erratum in
-
Publisher Correction: EMT cells increase breast cancer metastasis via paracrine GLI activation in neighbouring tumour cells.Nat Commun. 2018 Nov 12;9(1):4720. doi: 10.1038/s41467-018-07168-z. Nat Commun. 2018. PMID: 30420638 Free PMC article.
Abstract
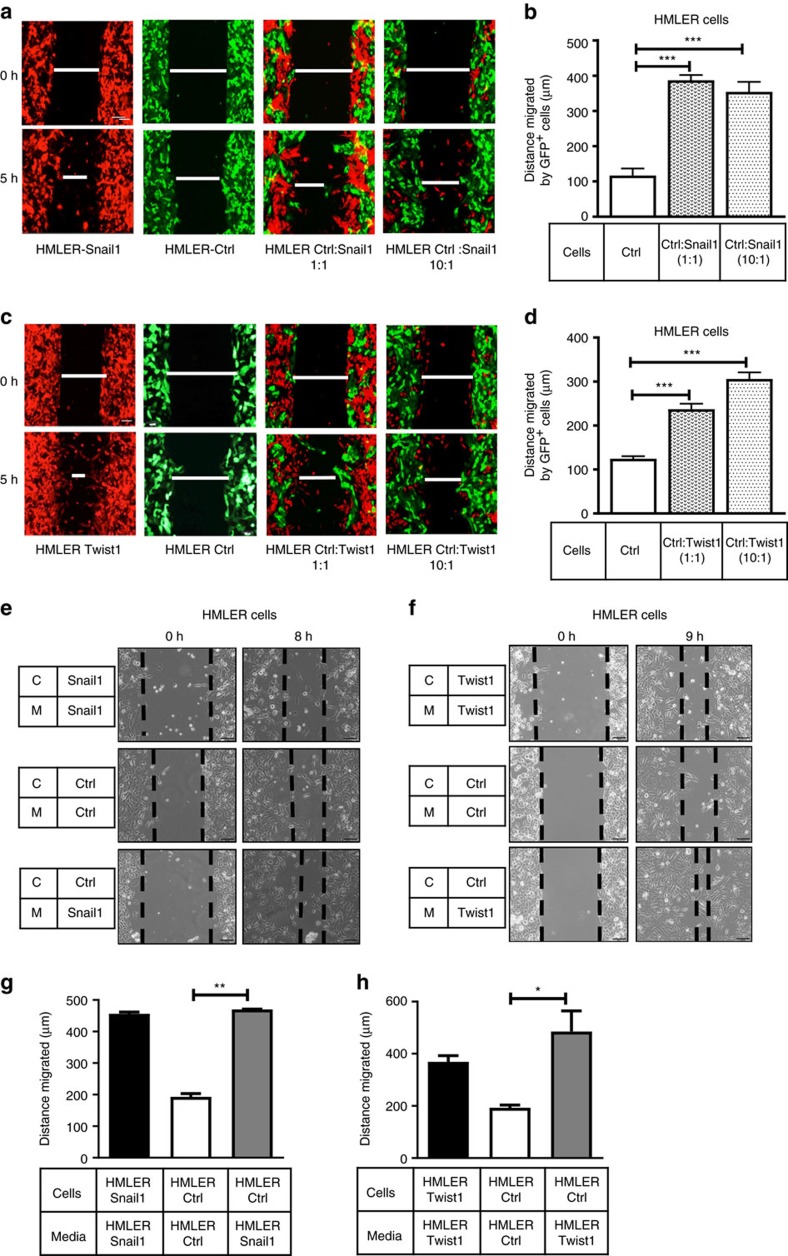
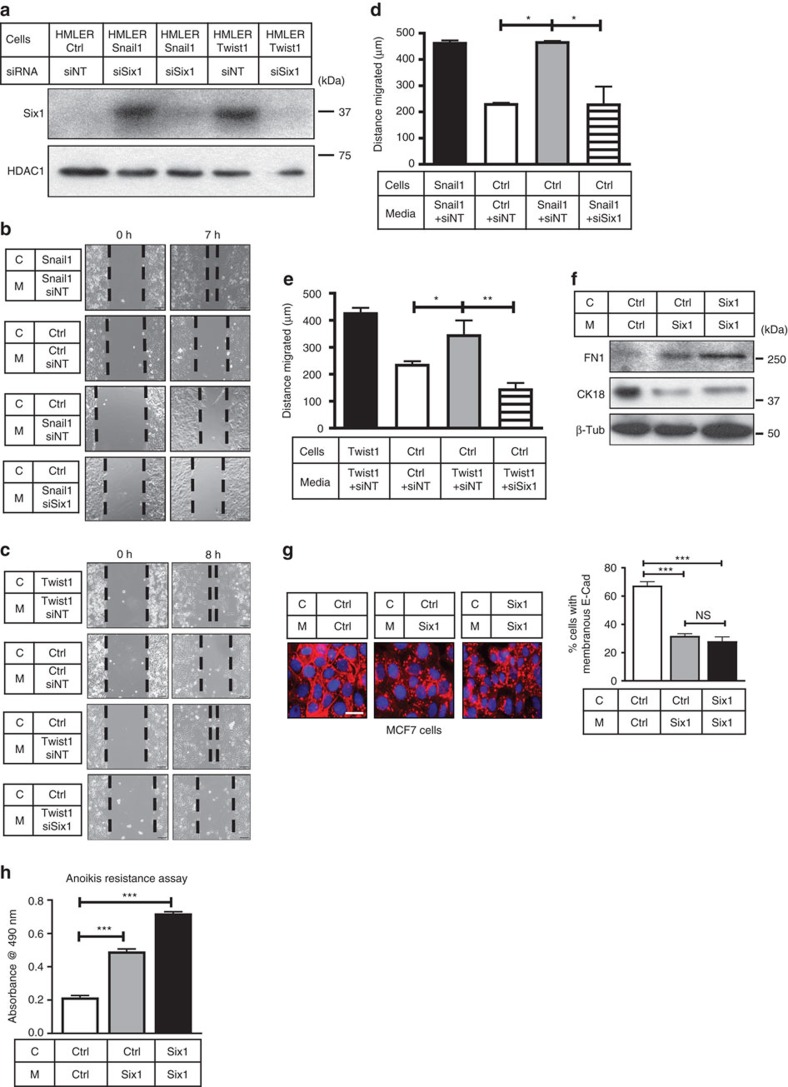
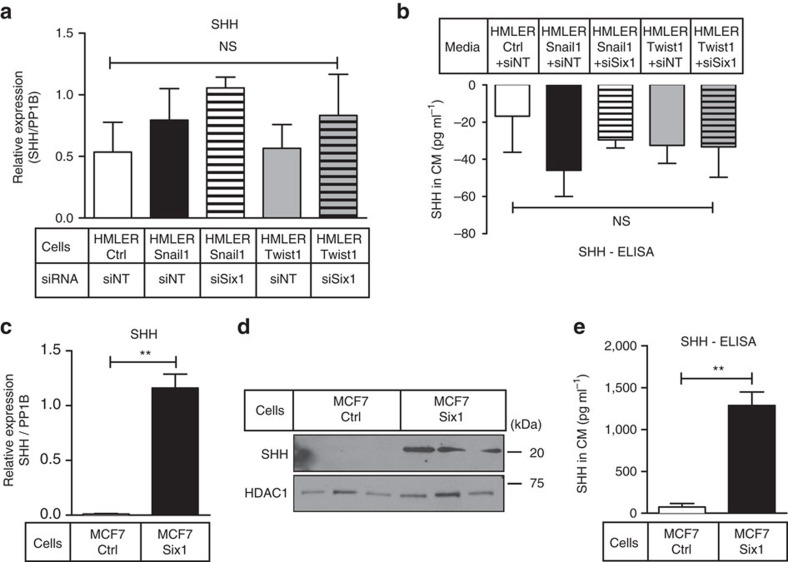
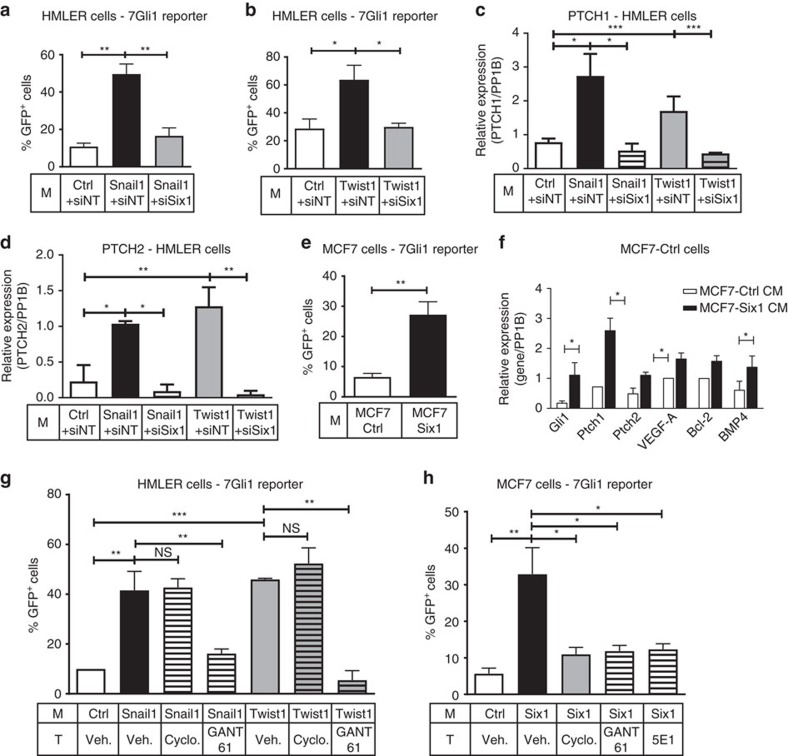
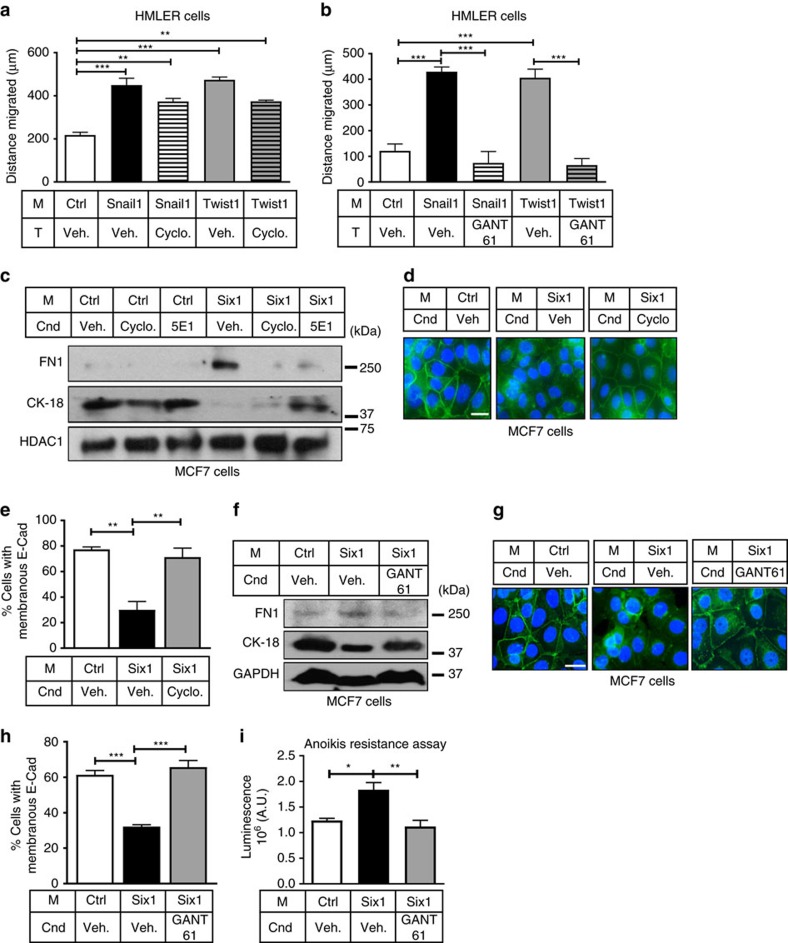
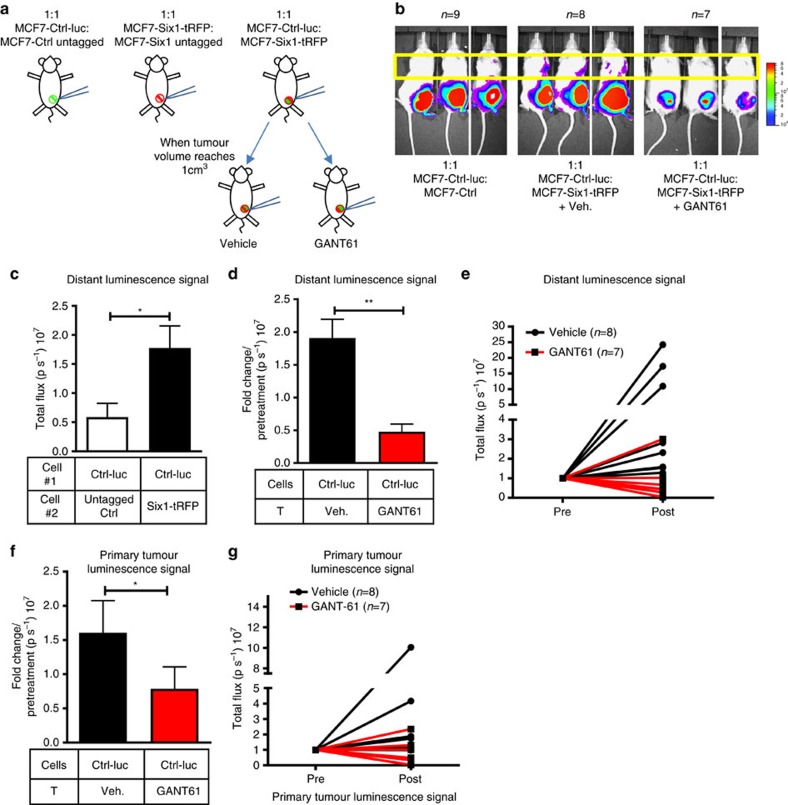
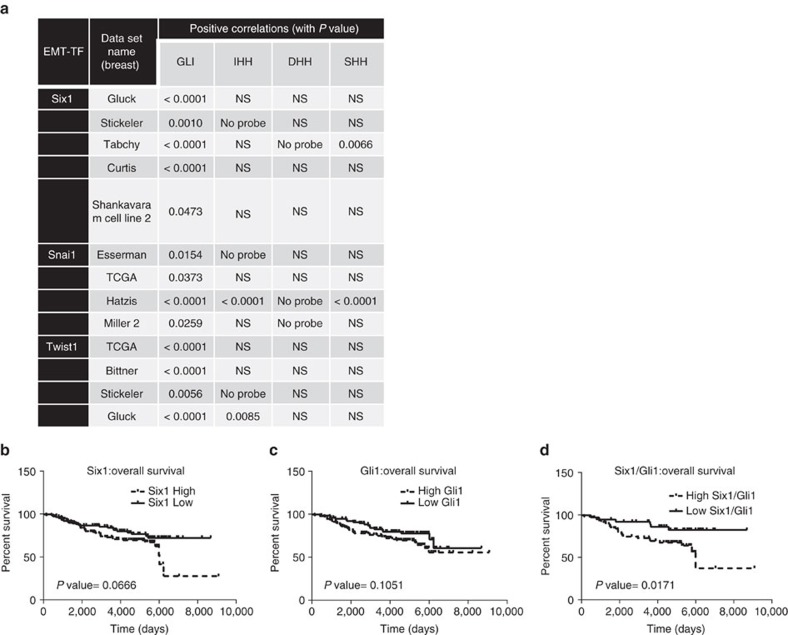
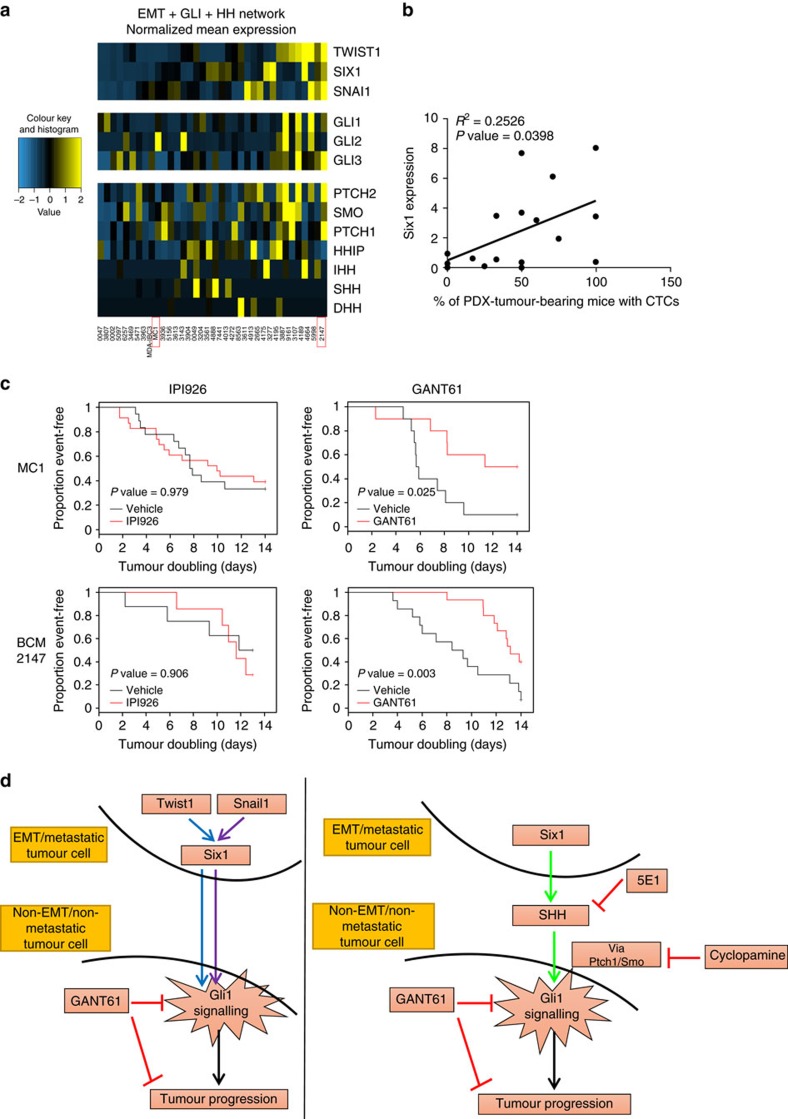
Recent fate-mapping studies concluded that EMT is not required for metastasis of carcinomas. Here we challenge this conclusion by showing that these studies failed to account for possible crosstalk between EMT and non-EMT cells that promotes dissemination of non-EMT cells. In breast cancer models, EMT cells induce increased metastasis of weakly metastatic, non-EMT tumour cells in a paracrine manner, in part by non-cell autonomous activation of the GLI transcription factor. Treatment with GANT61, a GLI1/2 inhibitor, but not with IPI 926, a Smoothened inhibitor, blocks this effect and inhibits growth in PDX models. In human breast tumours, the EMT-transcription factors strongly correlate with activated Hedgehog/GLI signalling but not with the Hh ligands. Our findings indicate that EMT contributes to metastasis via non-cell autonomous effects that activate the Hh pathway. Although all Hh inhibitors may act against tumours with canonical Hh/GLI signalling, only GLI inhibitors would act against non-canonical EMT-induced GLI activation.
Conflict of interest statement
M.T.L. is a manager in StemMed Holdings LP and a limited partner in StemMed Ltd. The remaining authors declare no competing financial interests.
Figures
References
-
- McGranahan N. & Swanton C. Biological and therapeutic impact of intratumour heterogeneity in cancer evolution. Cancer Cell 27, 15–26 (2015). - PubMed
-
- Thiery J. P., Acloque H., Huang R. Y. & Nieto M. A. Epithelial-mesenchymal transitions in development and disease. Cell 139, 871–890 (2009). - PubMed
-
- Gunasinghe N. P., Wells A., Thompson E. W. & Hugo H. J. Mesenchymal-epithelial transition (MET) as a mechanism for metastatic colonisation in breast cancer. Cancer Metastasis Rev. 31, 469–478 (2012). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
