

mTOR controls ChREBP transcriptional activity and pancreatic β cell survival under diabetic stress
- PMID: 28606928
- PMCID: PMC5496625
- DOI: 10.1083/jcb.201701085
mTOR controls ChREBP transcriptional activity and pancreatic β cell survival under diabetic stress
Abstract
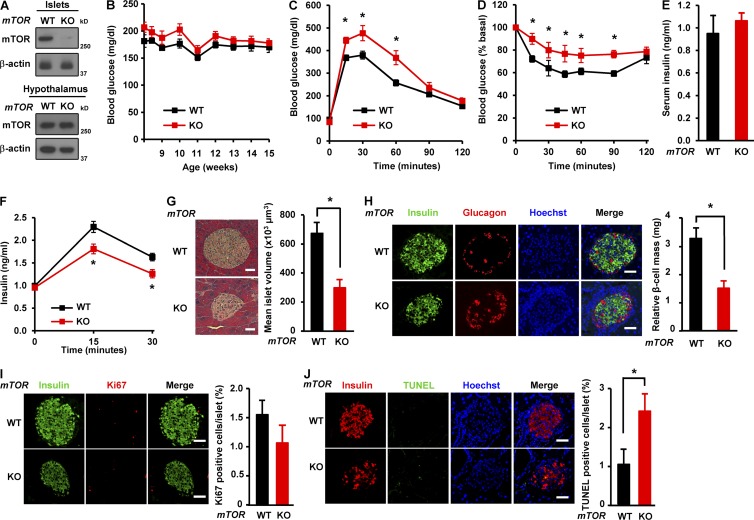
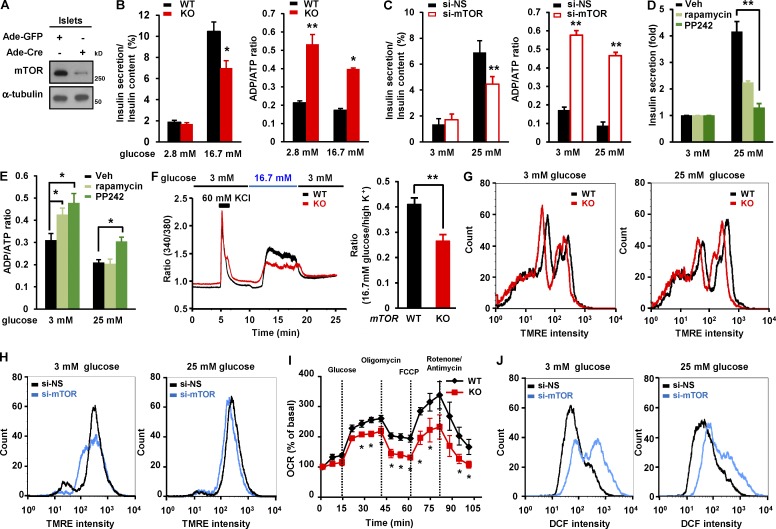
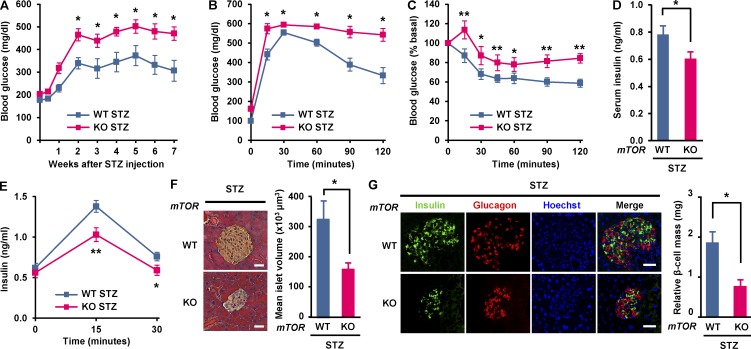
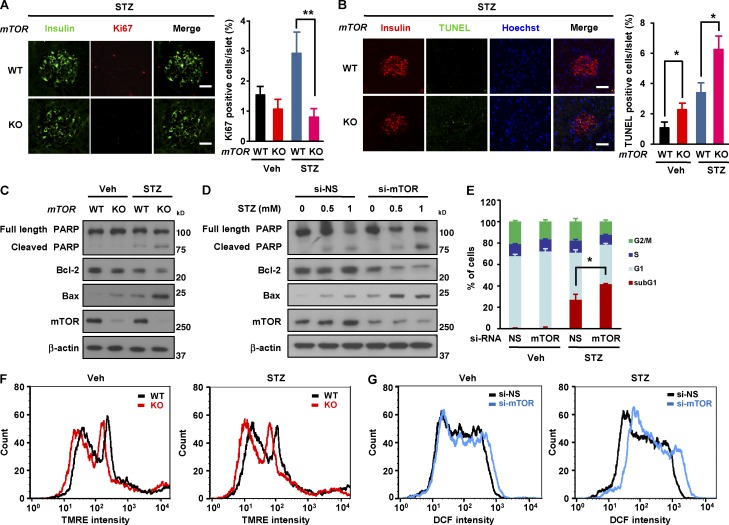
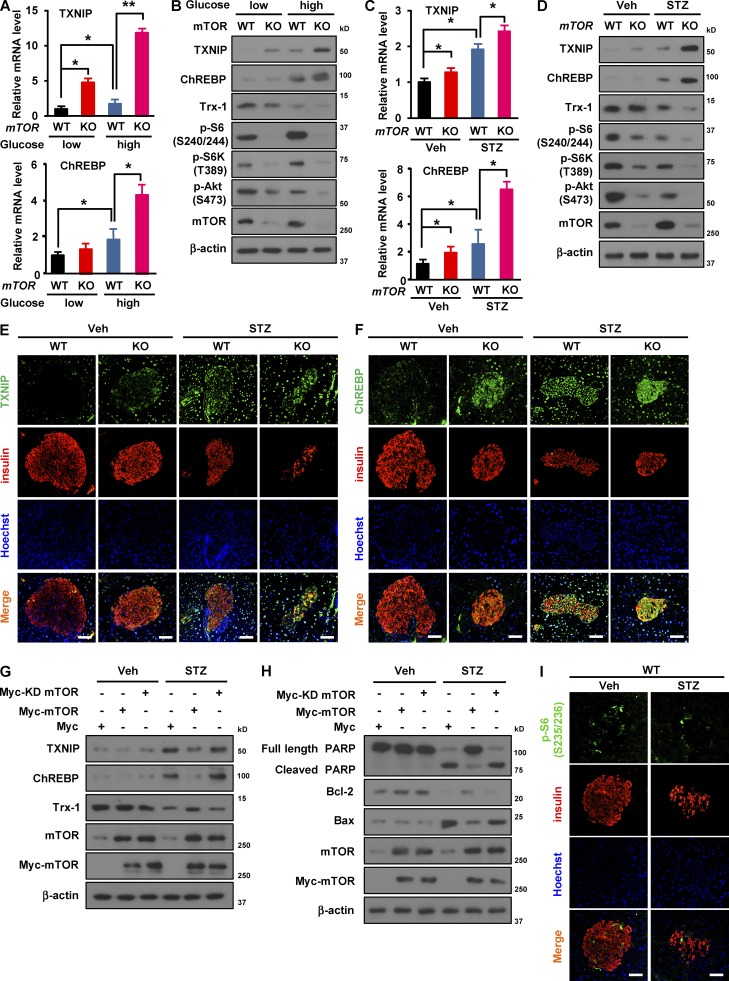
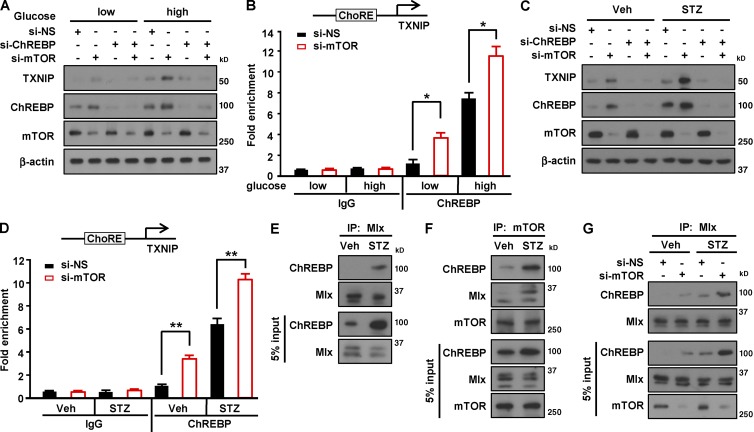
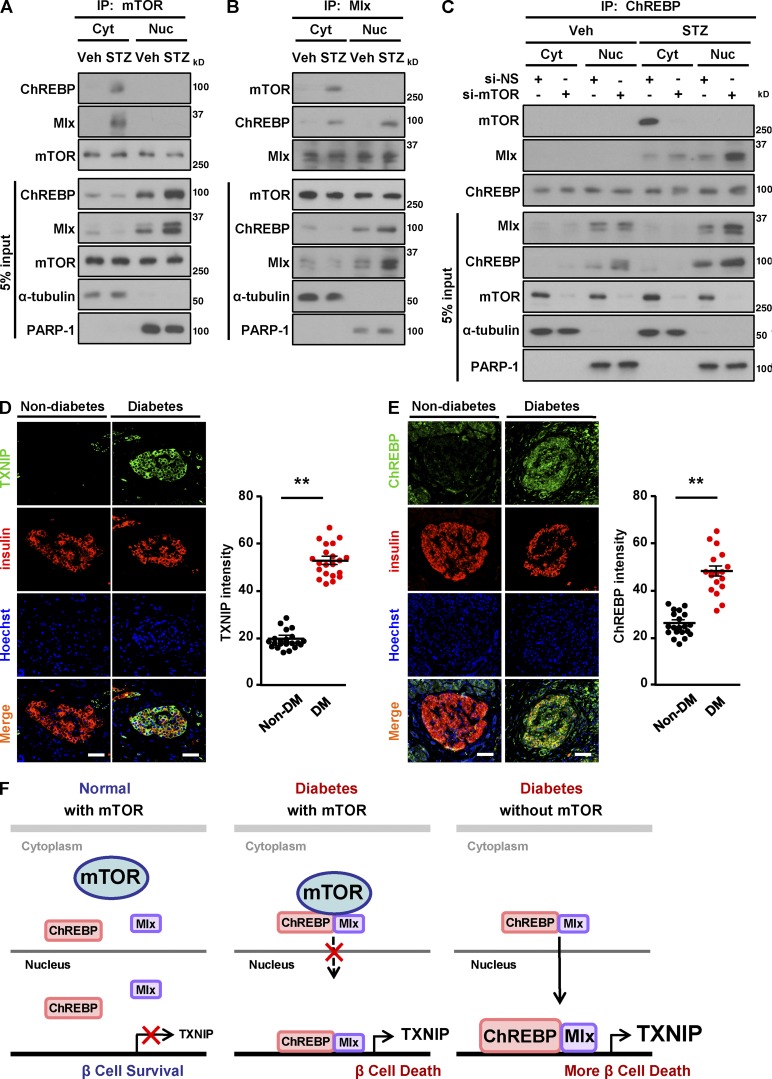
Impaired nutrient sensing and dysregulated glucose homeostasis are common in diabetes. However, how nutrient-sensitive signaling components control glucose homeostasis and β cell survival under diabetic stress is not well understood. Here, we show that mice lacking the core nutrient-sensitive signaling component mammalian target of rapamycin (mTOR) in β cells exhibit reduced β cell mass and smaller islets. mTOR deficiency leads to a severe reduction in β cell survival and increased mitochondrial oxidative stress in chemical-induced diabetes. Mechanistically, we find that mTOR associates with the carbohydrate-response element-binding protein (ChREBP)-Max-like protein complex and inhibits its transcriptional activity, leading to decreased expression of thioredoxin-interacting protein (TXNIP), a potent inducer of β cell death and oxidative stress. Consistent with this, the levels of TXNIP and ChREBP were highly elevated in human diabetic islets and mTOR-deficient mouse islets. Thus, our results suggest that a nutrient-sensitive mTOR-regulated transcriptional network could be a novel target to improve β cell survival and glucose homeostasis in diabetes.
© 2017 Chau et al.
Figures
References
-
- Bar-Peled L., Chantranupong L., Cherniack A.D., Chen W.W., Ottina K.A., Grabiner B.C., Spear E.D., Carter S.L., Meyerson M., and Sabatini D.M.. 2013. A Tumor suppressor complex with GAP activity for the Rag GTPases that signal amino acid sufficiency to mTORC1. Science. 340:1100–1106. 10.1126/science.1232044 - DOI - PMC - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
