

Autophagy downstream of endosomal Toll-like receptor signaling in macrophages is a key mechanism for resistance to Leishmania major infection
- PMID: 28607148
- PMCID: PMC5555173
- DOI: 10.1074/jbc.M117.780981
Autophagy downstream of endosomal Toll-like receptor signaling in macrophages is a key mechanism for resistance to Leishmania major infection
Abstract
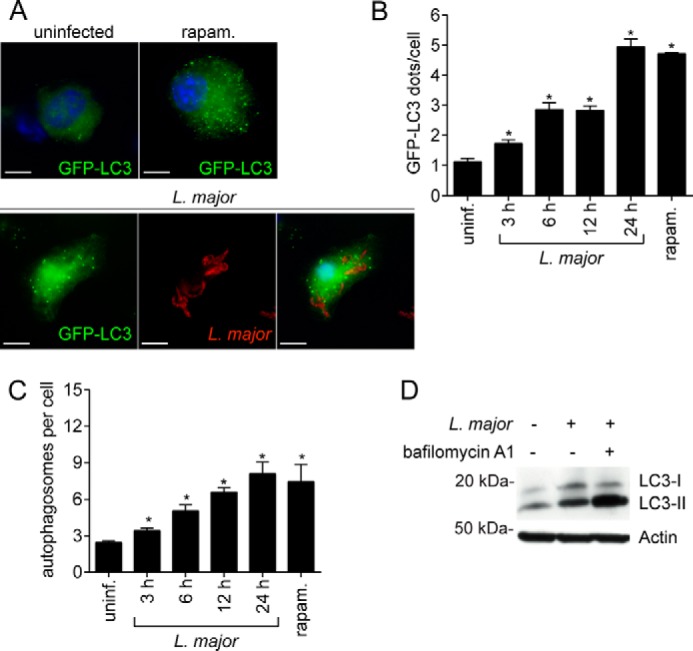
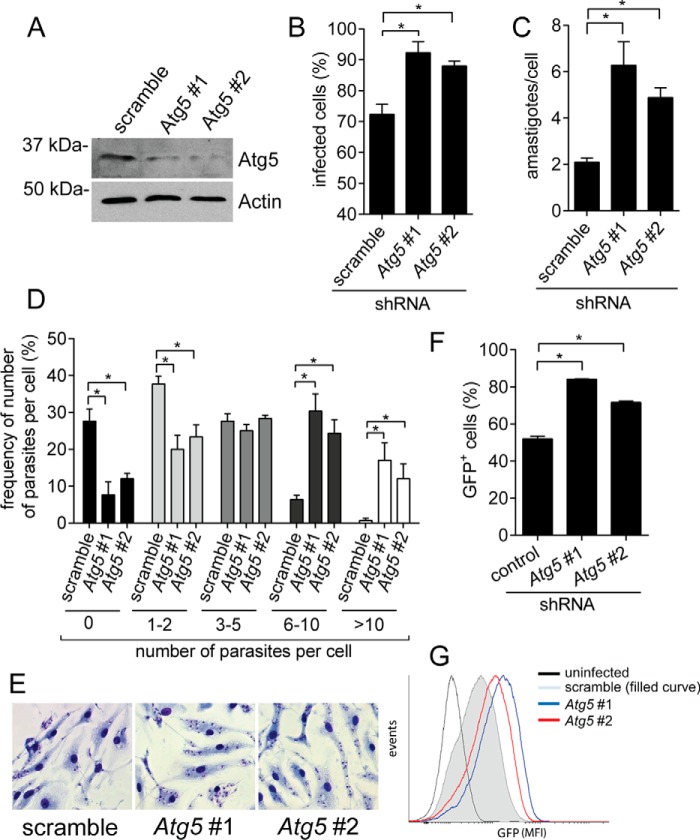
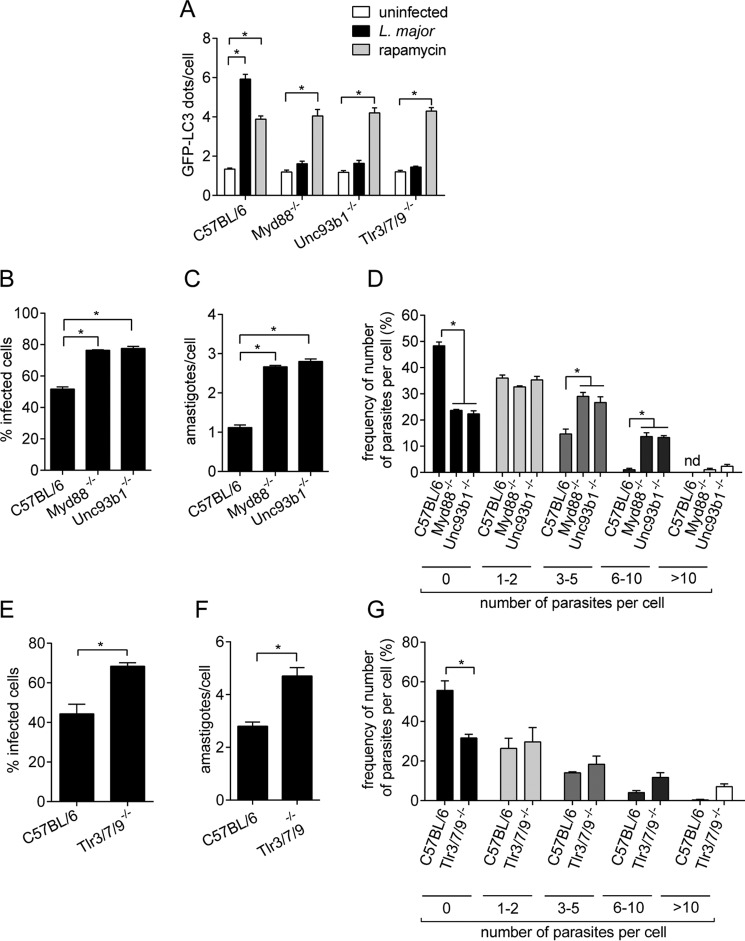
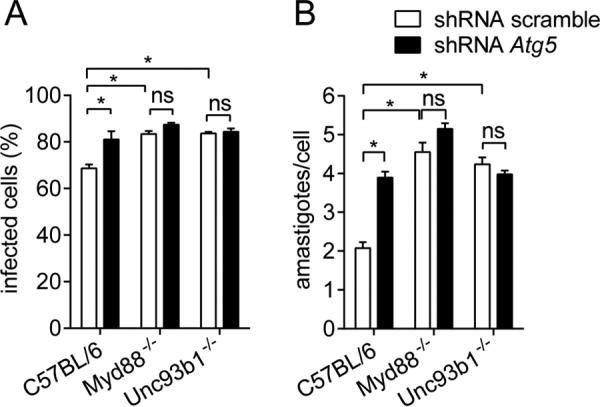
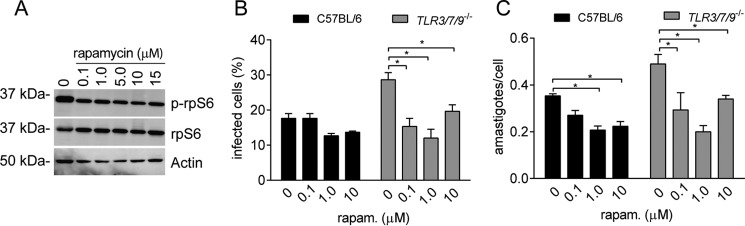
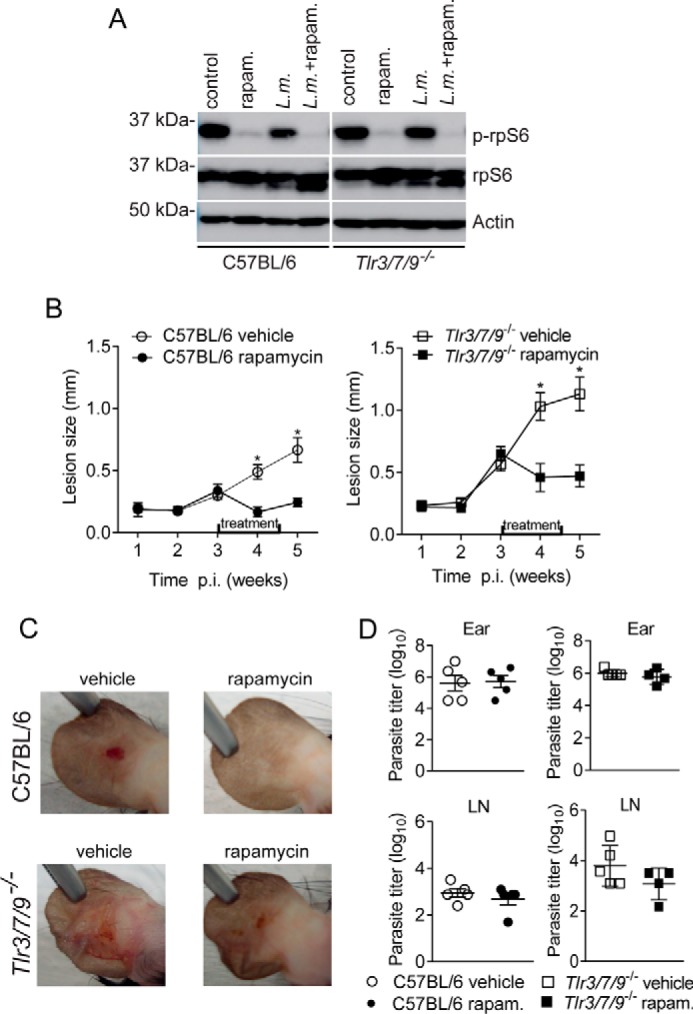
Leishmaniasis is caused by protozoan parasites of the genus Leishmania In mammalians, these parasites survive and replicate in macrophages and parasite elimination by macrophages is critical for host resistance. Endosomal Toll-like receptors (TLRs) have been shown to be crucial for resistance to Leishmania major in vivo For example, mice in the resistant C57BL/6 genetic background that are triple-deficient for TLR3, -7, and -9 (Tlr3/7/9-/-) are highly susceptible to L. major infection. Tlr3/7/9-/- mice are as susceptible as mice deficient in MyD88 or UNC93B1, a chaperone required for appropriate localization of endosomal TLRs, but the mechanisms are unknown. Here we found that macrophages infected with L. major undergo autophagy, which effectively accounted for restriction of parasite replication. Signaling via endosomal TLRs was required for autophagy because macrophages deficient for TLR3, -7, and 9, UNC93B1, or MyD88 failed to undergo L. major-induced autophagy. We also confirmed that Myd88-/-, Tlr3/7/9-/-, and Unc93b1-/- cells were highly permissive to L. major replication. Accordingly, shRNA-mediated suppression of Atg5, an E3 ubiquitin ligase essential for autophagosome elongation, in macrophages impaired the restriction of L. major replication in C57BL/6, but did not affect parasite replication in Myd88-/- or Unc93b1-/- macrophages. Rapamycin treatment reduced inflammatory lesions formed in the ears of Leishmania-infected C57BL/6 and Tlr3/7/9-/- mice, indicating that autophagy operates downstream of TLR signaling and is relevant for disease development in vivo Collectively, our results indicate that autophagy contributes to macrophage resistance to L. major replication, and mechanistically explain the previously described endosomal TLR-mediated resistance to L. major infection.
Keywords: Leishmania; autophagy; infection; macrophage; toll-like receptor (TLR).
© 2017 by The American Society for Biochemistry and Molecular Biology, Inc.
Conflict of interest statement
The authors declare that they have no conflicts of interest with the contents of this article
Figures
Similar articles
-
UNC93B1 and nucleic acid-sensing Toll-like receptors mediate host resistance to infection with Leishmania major.J Biol Chem. 2013 Mar 8;288(10):7127-36. doi: 10.1074/jbc.M112.407684. Epub 2013 Jan 16. J Biol Chem. 2013. PMID: 23325805 Free PMC article.
-
Release from UNC93B1 reinforces the compartmentalized activation of select TLRs.Nature. 2019 Nov;575(7782):371-374. doi: 10.1038/s41586-019-1611-7. Epub 2019 Sep 23. Nature. 2019. PMID: 31546247 Free PMC article.
-
Transcriptomic Analysis and C-Terminal Epitope Tagging Reveal Differential Processing and Signaling of Endogenous TLR3 and TLR7.Front Immunol. 2021 Jun 15;12:686060. doi: 10.3389/fimmu.2021.686060. eCollection 2021. Front Immunol. 2021. PMID: 34211474 Free PMC article.
-
Cell Surface Expression of Endosomal Toll-Like Receptors-A Necessity or a Superfluous Duplication?Front Immunol. 2021 Feb 1;11:620972. doi: 10.3389/fimmu.2020.620972. eCollection 2020. Front Immunol. 2021. PMID: 33597952 Free PMC article. Review.
-
An unexpected role for RNA-sensing toll-like receptors in a murine model of DNA accrual.Clin Exp Rheumatol. 2015 Jul-Aug;33(4 Suppl 92):S70-3. Epub 2015 Oct 12. Clin Exp Rheumatol. 2015. PMID: 26457825 Free PMC article. Review.
Cited by
-
Leishmania RNA virus exacerbates Leishmaniasis by subverting innate immunity via TLR3-mediated NLRP3 inflammasome inhibition.Nat Commun. 2019 Nov 21;10(1):5273. doi: 10.1038/s41467-019-13356-2. Nat Commun. 2019. PMID: 31754185 Free PMC article.
-
Autophagy in protists and their hosts: When, how and why?Autophagy Rep. 2023;2(1):2149211. doi: 10.1080/27694127.2022.2149211. Epub 2023 Mar 9. Autophagy Rep. 2023. PMID: 37064813 Free PMC article.
-
Deciphering the Dual Role of Heligmosomoides polygyrus Antigens in Macrophage Modulation and Breast Cancer Cell Growth.Vet Sci. 2024 Feb 3;11(2):69. doi: 10.3390/vetsci11020069. Vet Sci. 2024. PMID: 38393087 Free PMC article.
-
Evidence of the Autophagic Process during the Fish Immune Response of Skeletal Muscle Cells against Piscirickettsia salmonis.Animals (Basel). 2023 Feb 28;13(5):880. doi: 10.3390/ani13050880. Animals (Basel). 2023. PMID: 36899738 Free PMC article.
-
TLR Specific Immune Responses against Helminth Infections.J Parasitol Res. 2017;2017:6865789. doi: 10.1155/2017/6865789. Epub 2017 Oct 31. J Parasitol Res. 2017. PMID: 29225962 Free PMC article. Review.
References
-
- O'Neill L. A., Golenbock D., and Bowie A. G. (2013) The history of Toll-like receptors: redefining innate immunity. Nat. Rev. Immunol. 13, 453–460 - PubMed
-
- de Veer M. J., Curtis J. M., Baldwin T. M., DiDonato J. A., Sexton A., McConville M. J., Handman E., and Schofield L. (2003) MyD88 is essential for clearance of Leishmania major: possible role for lipophosphoglycan and Toll-like receptor 2 signaling. Eur. J. Immunol. 33, 2822–2831 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
