

Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance
- PMID: 28607484
- PMCID: PMC5542814
- DOI: 10.1038/nature22794
Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance
Erratum in
-
Corrigendum: Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance.Nature. 2018 Mar 8;555(7695):274. doi: 10.1038/nature25162. Epub 2018 Feb 21. Nature. 2018. PMID: 29466340
Abstract
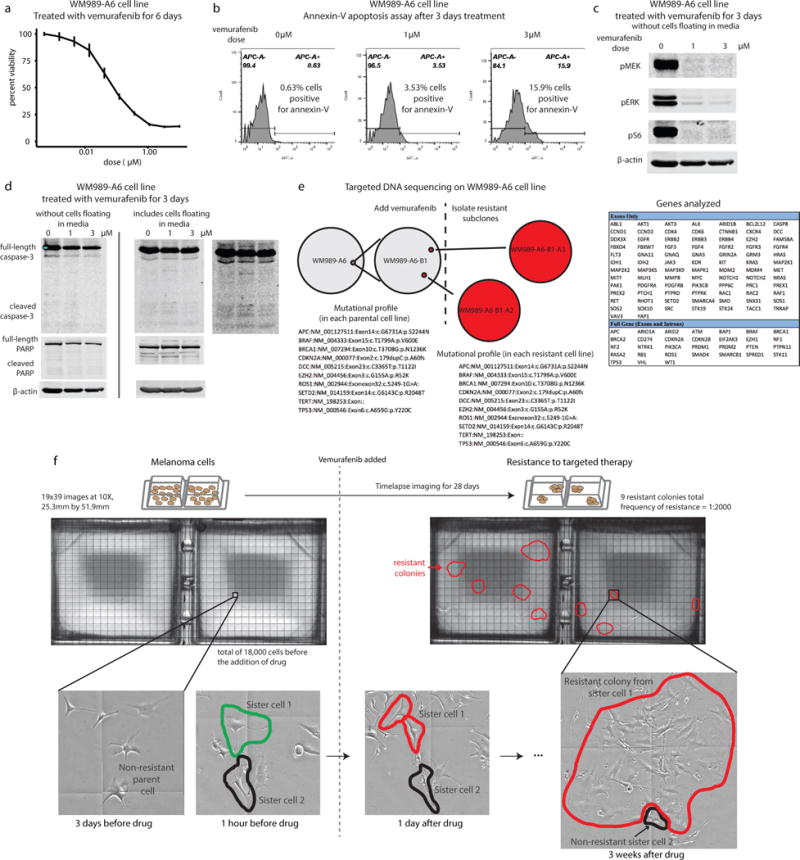
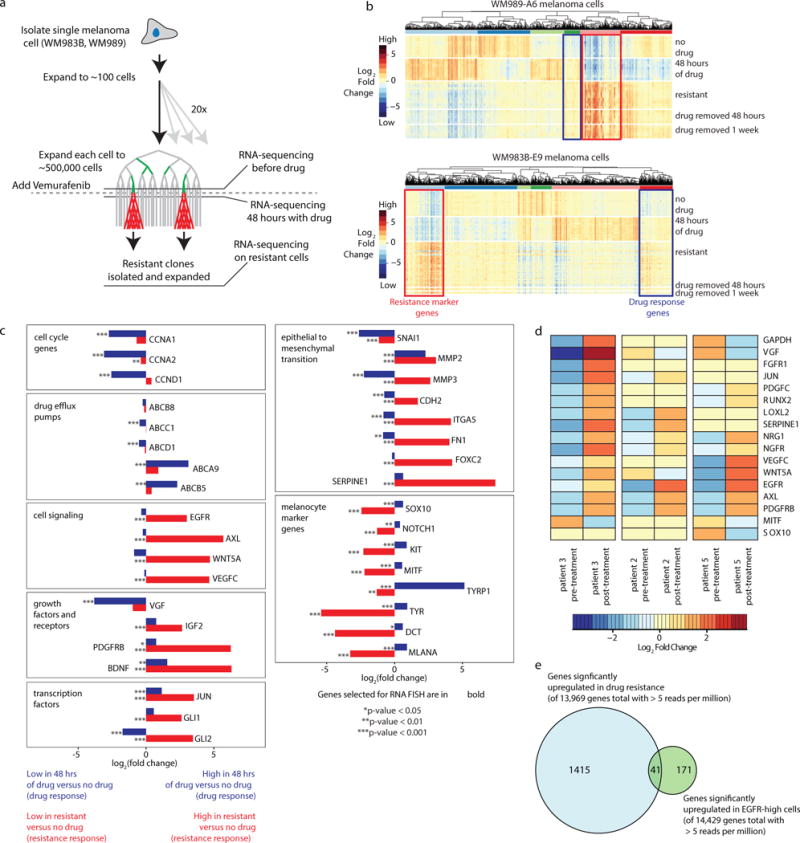
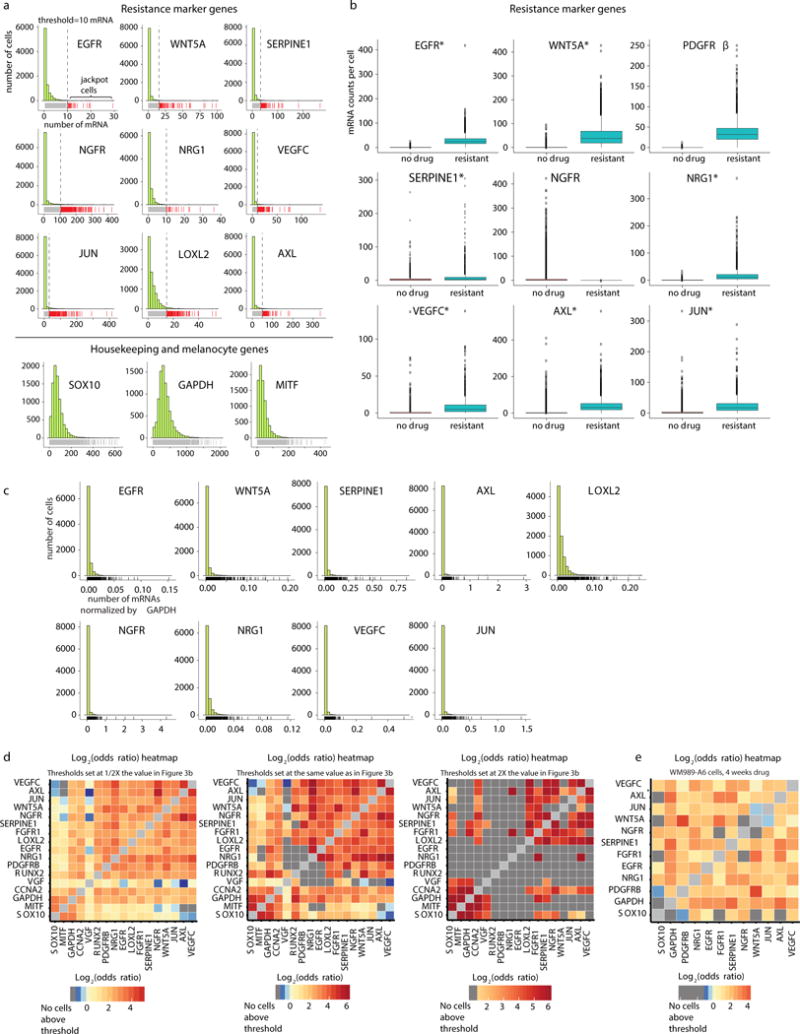
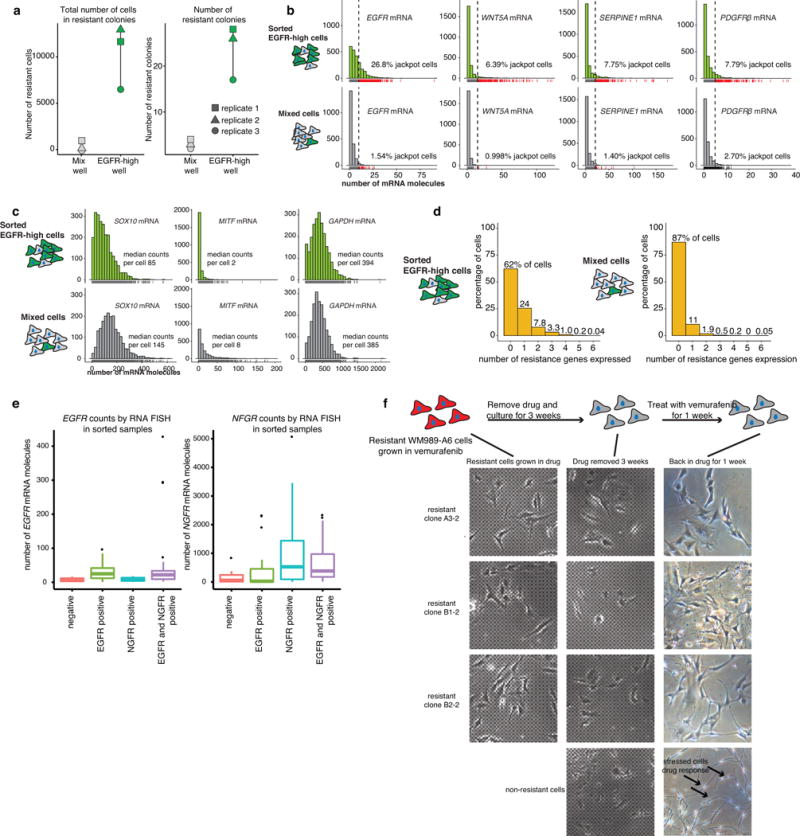
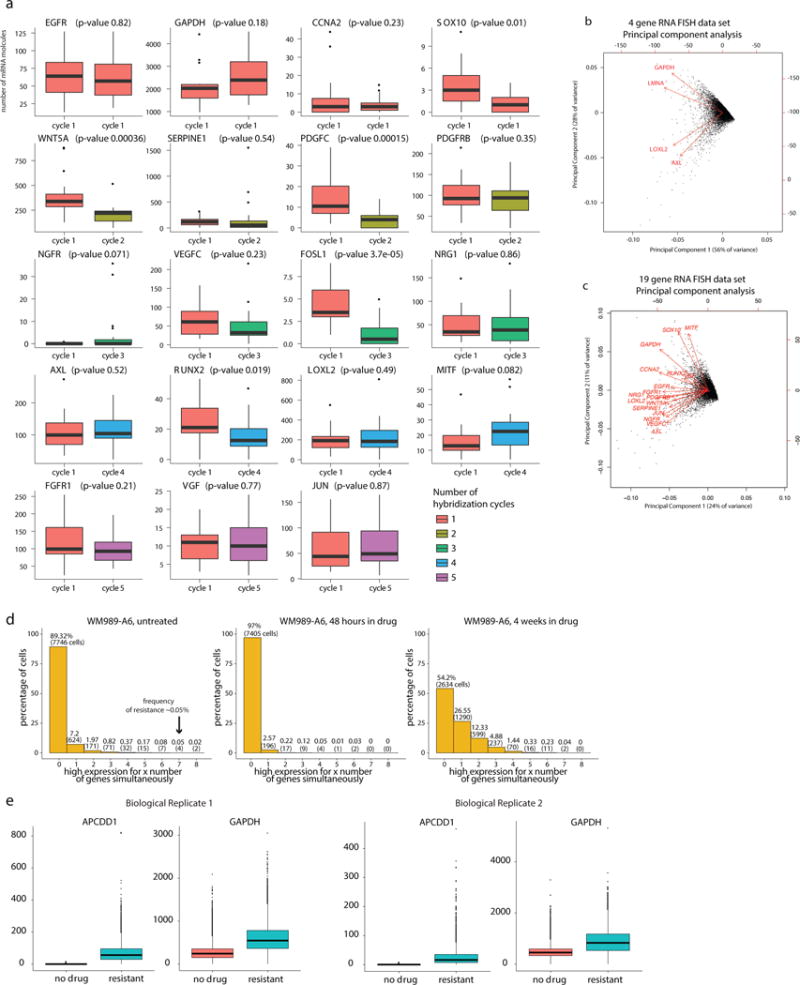
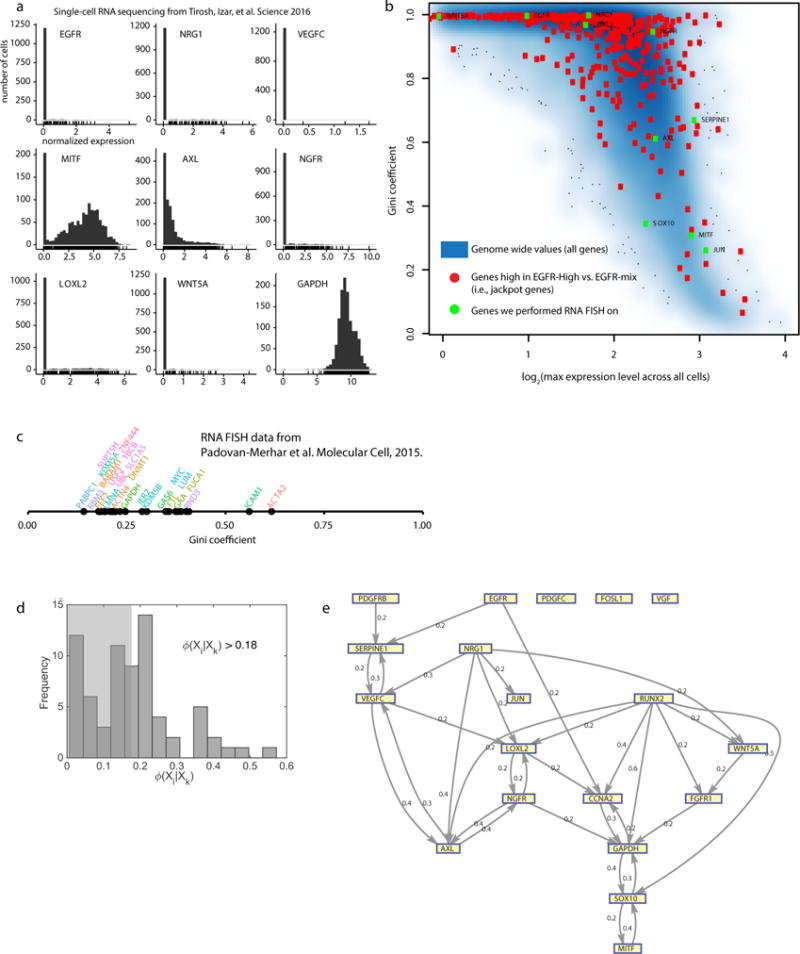
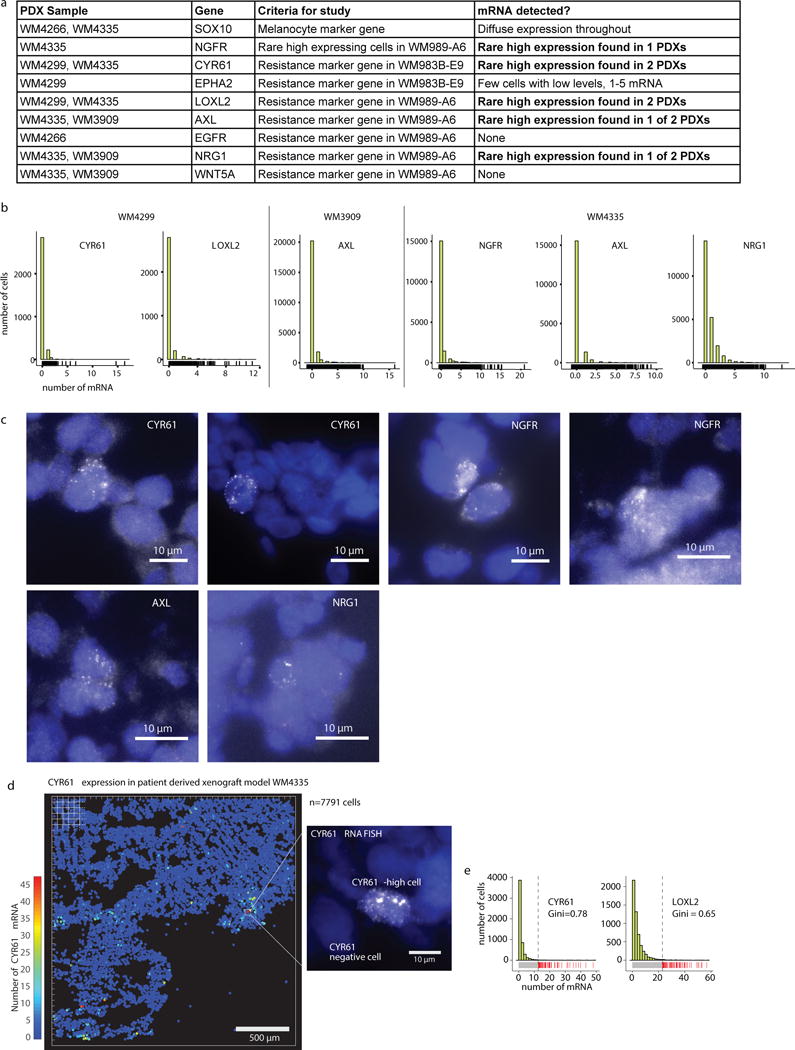
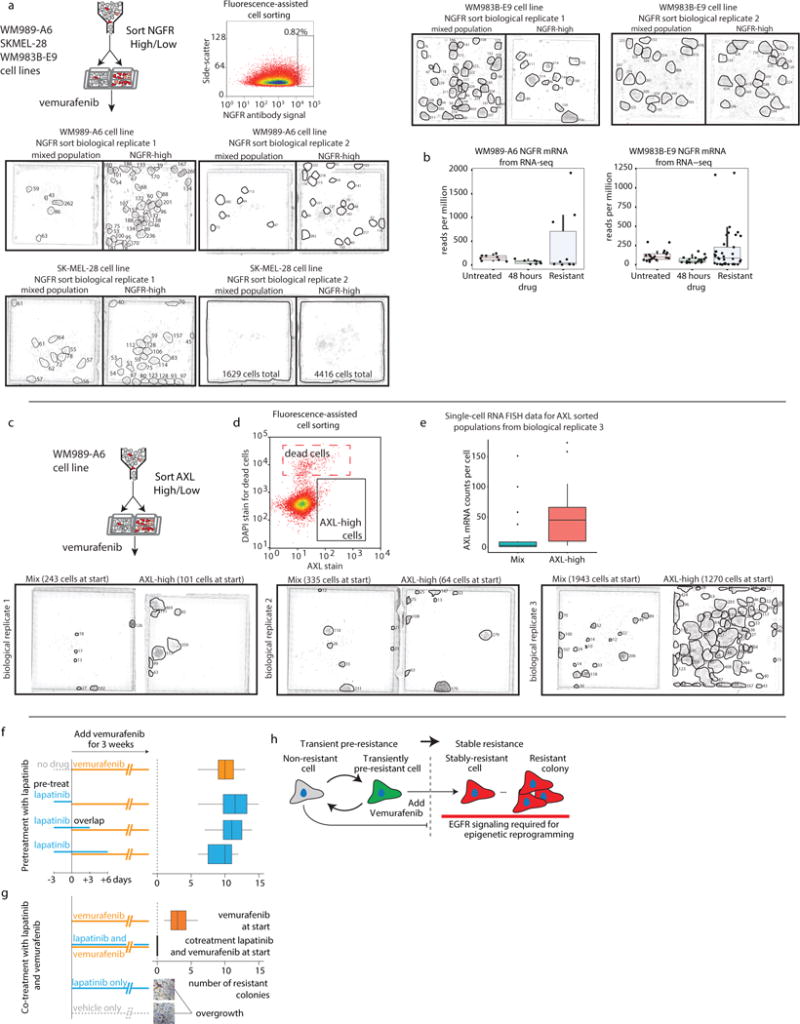
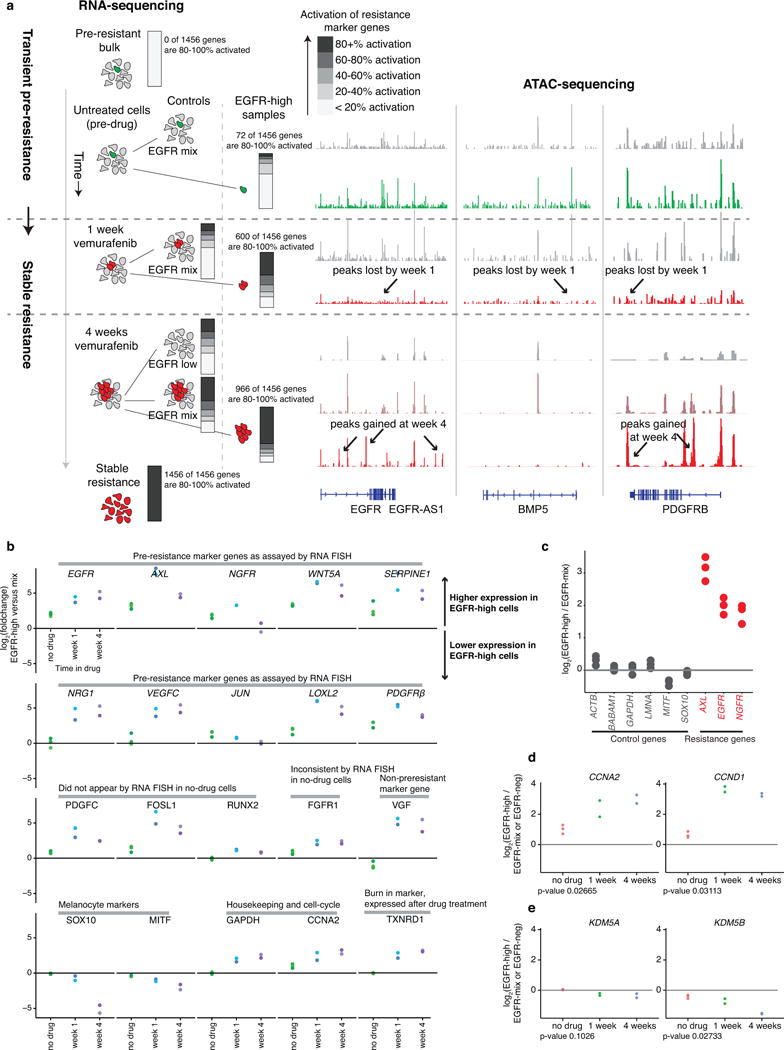
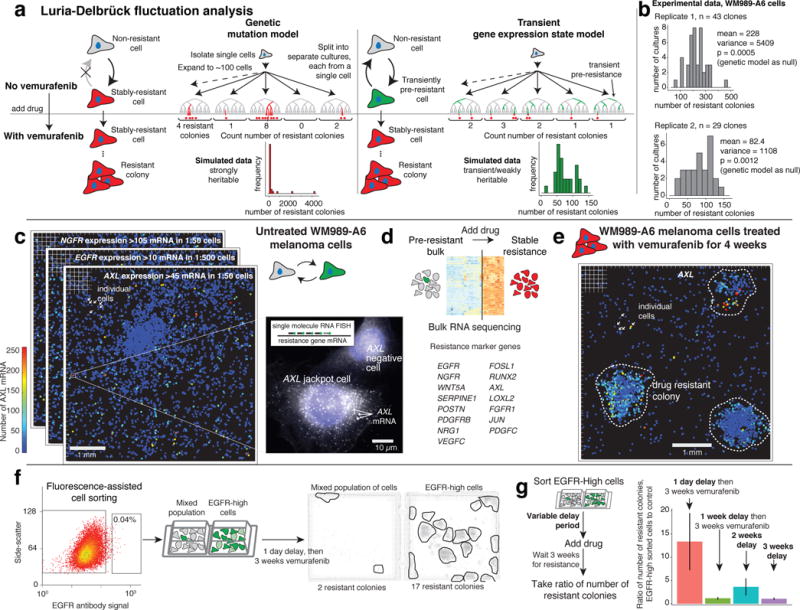
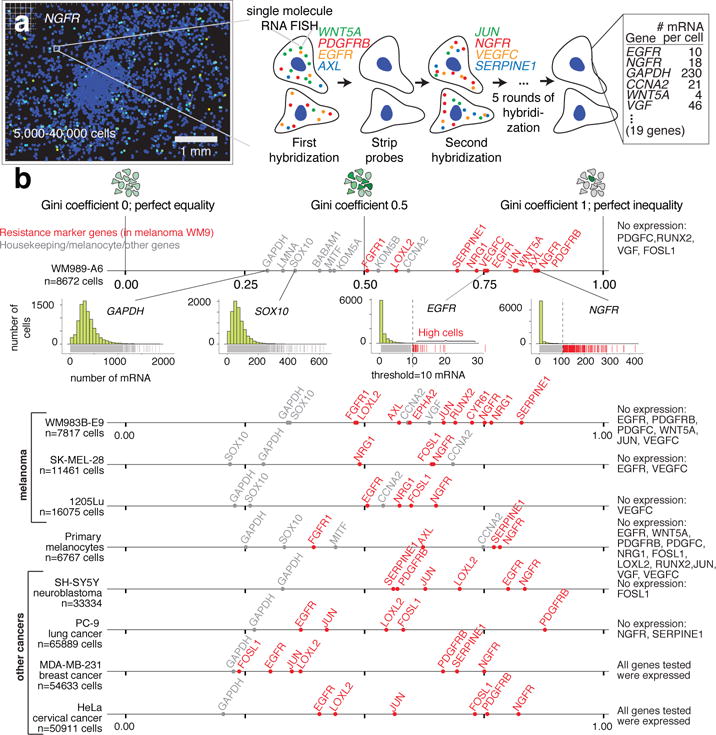
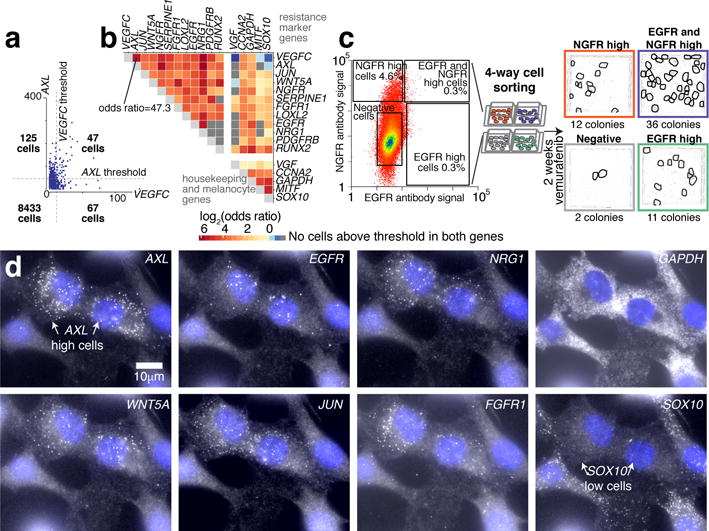
Therapies that target signalling molecules that are mutated in cancers can often have substantial short-term effects, but the emergence of resistant cancer cells is a major barrier to full cures. Resistance can result from secondary mutations, but in other cases there is no clear genetic cause, raising the possibility of non-genetic rare cell variability. Here we show that human melanoma cells can display profound transcriptional variability at the single-cell level that predicts which cells will ultimately resist drug treatment. This variability involves infrequent, semi-coordinated transcription of a number of resistance markers at high levels in a very small percentage of cells. The addition of drug then induces epigenetic reprogramming in these cells, converting the transient transcriptional state to a stably resistant state. This reprogramming begins with a loss of SOX10-mediated differentiation followed by activation of new signalling pathways, partially mediated by the activity of the transcription factors JUN and/or AP-1 and TEAD. Our work reveals the multistage nature of the acquisition of drug resistance and provides a framework for understanding resistance dynamics in single cells. We find that other cell types also exhibit sporadic expression of many of these same marker genes, suggesting the existence of a general program in which expression is displayed in rare subpopulations of cells.
Figures

Comment in
-
Epigenetics make transient states of cancer therapy resistance permanent.Sci Transl Med. 2017 Jun 28;9(396):eaan6729. doi: 10.1126/scitranslmed.aan6729. Sci Transl Med. 2017. PMID: 28659441
-
Therapeutic resistance: Transcribing patterns of resistance.Nat Rev Cancer. 2017 Jul 25;17(8):452-453. doi: 10.1038/nrc.2017.59. Nat Rev Cancer. 2017. PMID: 28740111 No abstract available.
-
Rare, yet relevant tumor cells - A new twist to melanoma cell plasticity.Pigment Cell Melanoma Res. 2018 Jan;31(1):7-9. doi: 10.1111/pcmr.12643. Epub 2017 Oct 3. Pigment Cell Melanoma Res. 2018. PMID: 28898550 No abstract available.
References
-
- Trunzer K, et al. Pharmacodynamic effects and mechanisms of resistance to vemurafenib in patients with metastatic melanoma. J Clin Oncol. 2013;31:1767–1774. - PubMed
-
- Garraway LA, Jänne PA. Circumventing cancer drug resistance in the era of personalized medicine. Cancer Discov. 2012;2:214–226. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
