

Population genomics and the evolution of virulence in the fungal pathogen Cryptococcus neoformans
- PMID: 28611159
- PMCID: PMC5495072
- DOI: 10.1101/gr.218727.116
Population genomics and the evolution of virulence in the fungal pathogen Cryptococcus neoformans
Abstract
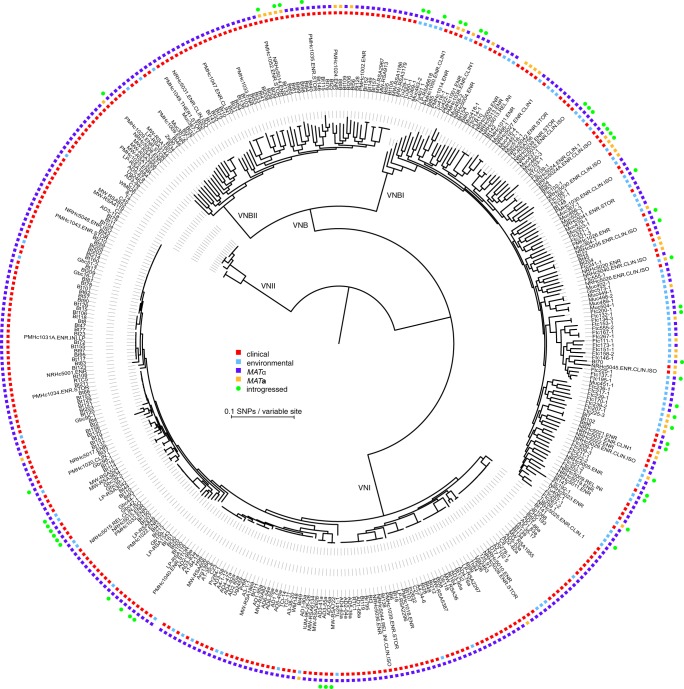
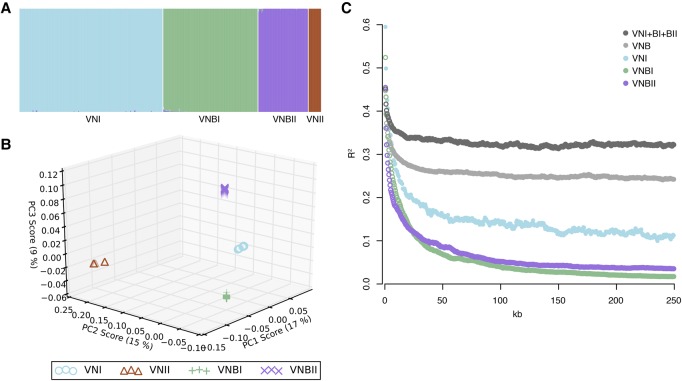
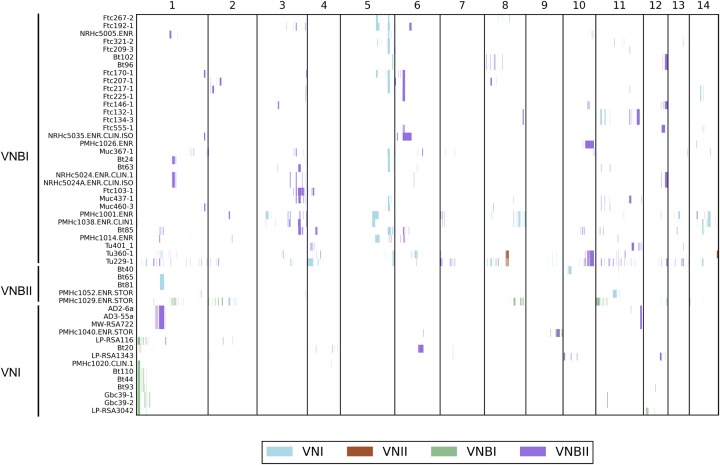
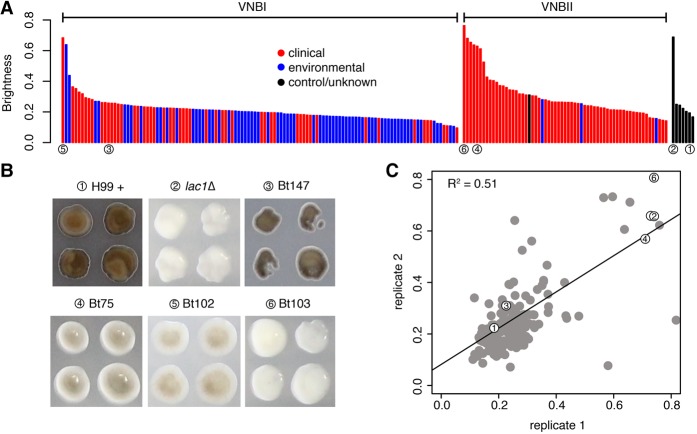
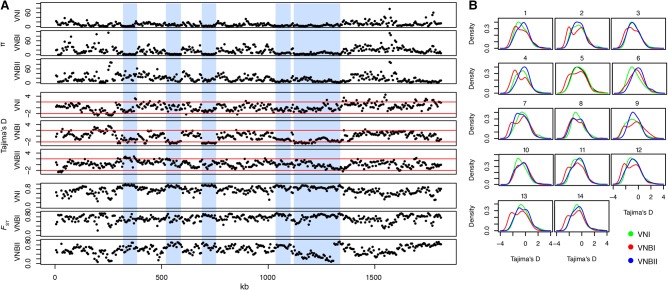
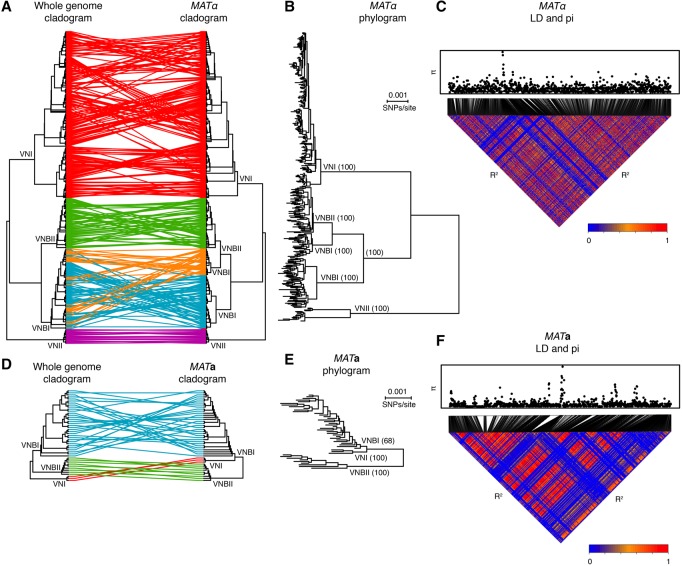
Cryptococcus neoformans is an opportunistic fungal pathogen that causes approximately 625,000 deaths per year from nervous system infections. Here, we leveraged a unique, genetically diverse population of C. neoformans from sub-Saharan Africa, commonly isolated from mopane trees, to determine how selective pressures in the environment coincidentally adapted C. neoformans for human virulence. Genome sequencing and phylogenetic analysis of 387 isolates, representing the global VNI and African VNB lineages, highlighted a deep, nonrecombining split in VNB (herein, VNBI and VNBII). VNBII was enriched for clinical samples relative to VNBI, while phenotypic profiling of 183 isolates demonstrated that VNBI isolates were significantly more resistant to oxidative stress and more heavily melanized than VNBII isolates. Lack of melanization in both lineages was associated with loss-of-function mutations in the BZP4 transcription factor. A genome-wide association study across all VNB isolates revealed sequence differences between clinical and environmental isolates in virulence factors and stress response genes. Inositol transporters and catabolism genes, which process sugars present in plants and the human nervous system, were identified as targets of selection in all three lineages. Further phylogenetic and population genomic analyses revealed extensive loss of genetic diversity in VNBI, suggestive of a history of population bottlenecks, along with unique evolutionary trajectories for mating type loci. These data highlight the complex evolutionary interplay between adaptation to natural environments and opportunistic infections, and that selection on specific pathways may predispose isolates to human virulence.
© 2017 Desjardins et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
References
-
- Benjamini Y, Hochberg Y. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B Methodol 57: 289–300.
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases