

Quantitative Analysis of the DNA Methylation Sensitivity of Transcription Factor Complexes
- PMID: 28614722
- PMCID: PMC5533174
- DOI: 10.1016/j.celrep.2017.05.069
Quantitative Analysis of the DNA Methylation Sensitivity of Transcription Factor Complexes
Abstract
Although DNA modifications play an important role in gene regulation, the underlying mechanisms remain elusive. We developed EpiSELEX-seq to probe the sensitivity of transcription factor binding to DNA modification in vitro using massively parallel sequencing. Feature-based modeling quantifies the effect of cytosine methylation (5mC) on binding free energy in a position-specific manner. Application to the human bZIP proteins ATF4 and C/EBPβ and three different Pbx-Hox complexes shows that 5mCpG can both increase and decrease affinity, depending on where the modification occurs within the protein-DNA interface. The TF paralogs tested vary in their methylation sensitivity, for which we provide a structural rationale. We show that 5mCpG can also enhance in vitro p53 binding and provide evidence for increased in vivo p53 occupancy at methylated binding sites, correlating with primed enhancer histone marks. Our results establish a powerful strategy for dissecting the epigenomic modulation of protein-DNA interactions and their role in gene regulation.
Keywords: 5-methyl-cytosine; ChIP-seq data; SELEX-seq; bZIP; basic leucine zipper proteins; bisulfite sequencing; epigenetic DNA modification; epigenomics; high-throughput in vitro protein-DNA interaction profiling; human Hox protein complexes; integrative analysis; methylome; transcription factors; tumor suppressor protein p53.
Copyright © 2017 The Author(s). Published by Elsevier Inc. All rights reserved.
Figures



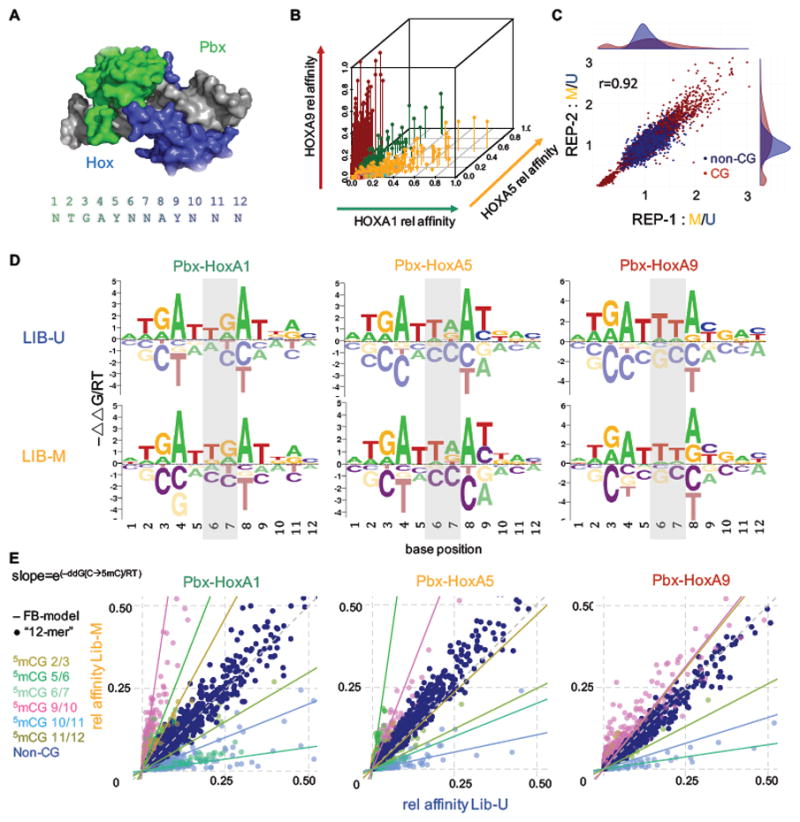
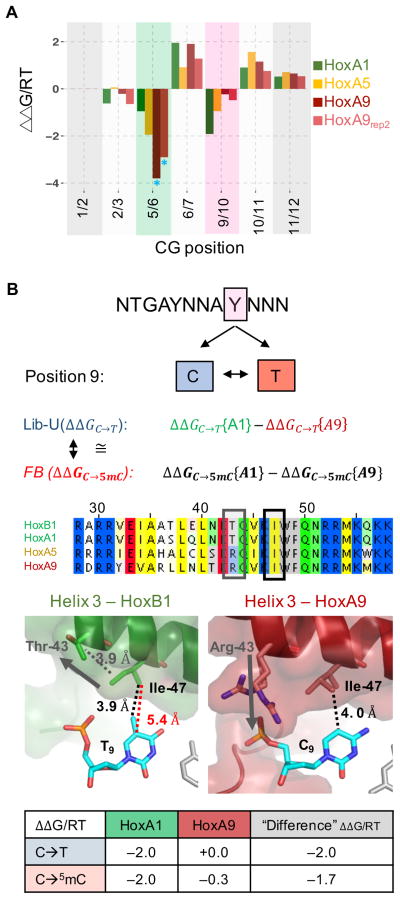

References
-
- Ambrosi C, Manzo M, Baubec T. Dynamics and Context-Dependent Roles of DNA Methylation. J Mol Biol 2017 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
