

Acamprosate in a mouse model of fragile X syndrome: modulation of spontaneous cortical activity, ERK1/2 activation, locomotor behavior, and anxiety
- PMID: 28616095
- PMCID: PMC5467053
- DOI: 10.1186/s11689-017-9184-y
Acamprosate in a mouse model of fragile X syndrome: modulation of spontaneous cortical activity, ERK1/2 activation, locomotor behavior, and anxiety
Abstract
Background: Fragile X Syndrome (FXS) occurs as a result of a silenced fragile X mental retardation 1 gene (FMR1) and subsequent loss of fragile X mental retardation protein (FMRP) expression. Loss of FMRP alters excitatory/inhibitory signaling balance, leading to increased neuronal hyperexcitability and altered behavior. Acamprosate (the calcium salt of N-acetylhomotaurinate), a drug FDA-approved for relapse prevention in the treatment of alcohol dependence in adults, is a novel agent with multiple mechanisms that may be beneficial for people with FXS. There are questions regarding the neuroactive effects of acamprosate and the significance of the molecule's calcium moiety. Therefore, the electrophysiological, cellular, molecular, and behavioral effects of acamprosate were assessed in the Fmr1-/y (knock out; KO) mouse model of FXS controlling for the calcium salt in several experiments.
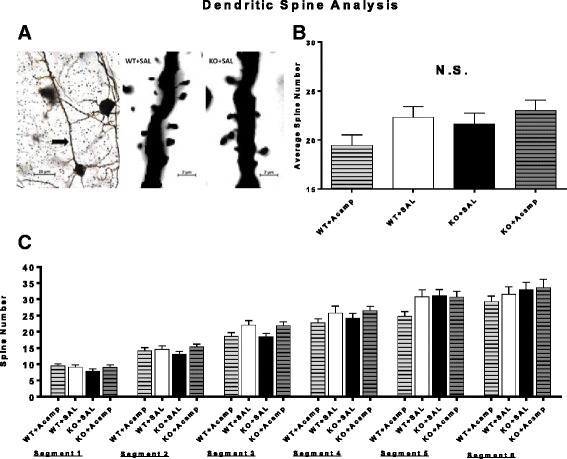
Methods: Fmr1 KO mice and their wild-type (WT) littermates were utilized to assess acamprosate treatment on cortical UP state parameters, dendritic spine density, and seizure susceptibility. Brain extracellular-signal regulated kinase 1/2 (ERK1/2) activation was used to investigate this signaling molecule as a potential biomarker for treatment response. Additional adult mice were used to assess chronic acamprosate treatment and any potential effects of the calcium moiety using CaCl2 treatment on behavior and nuclear ERK1/2 activation.
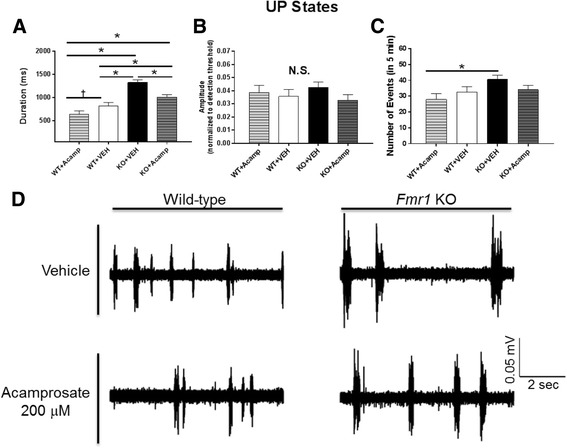
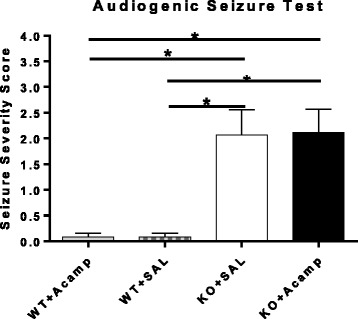
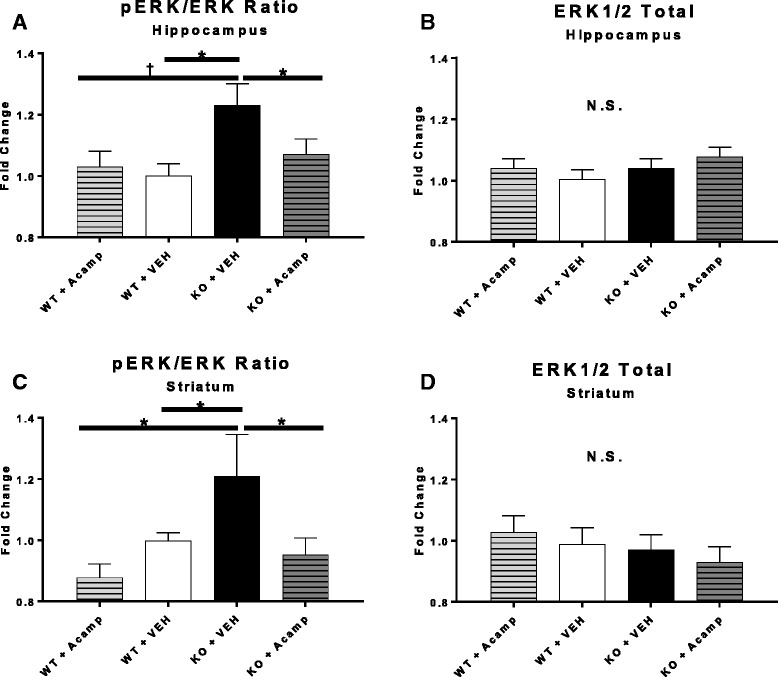
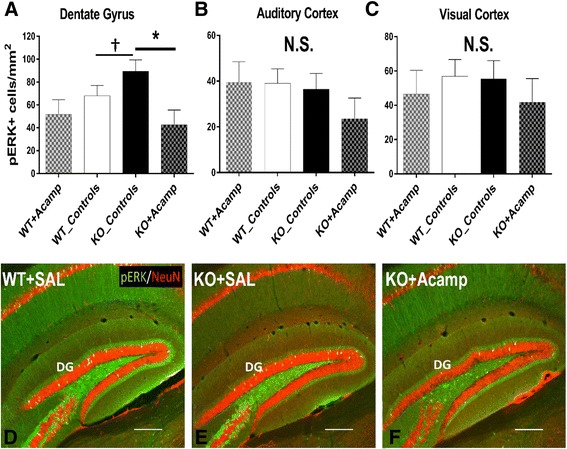
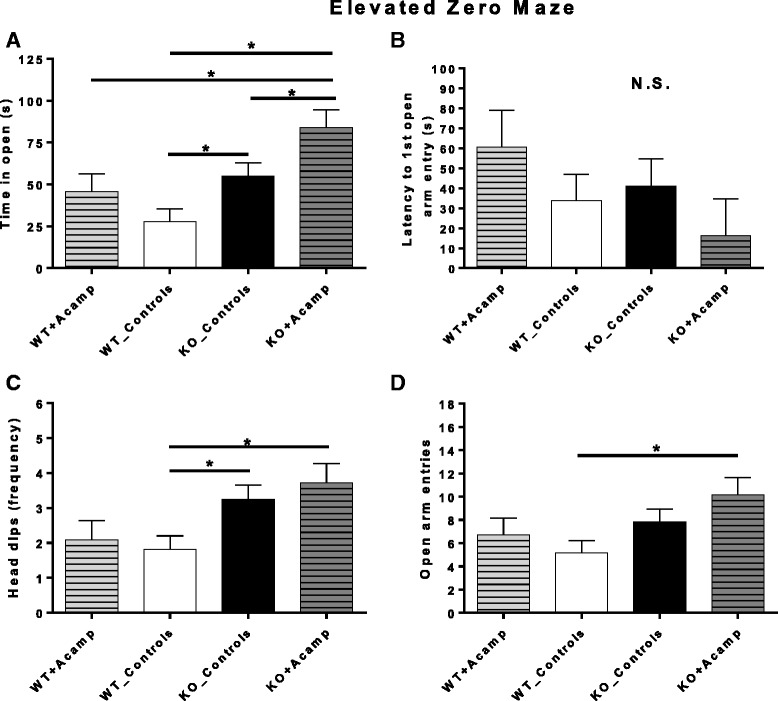
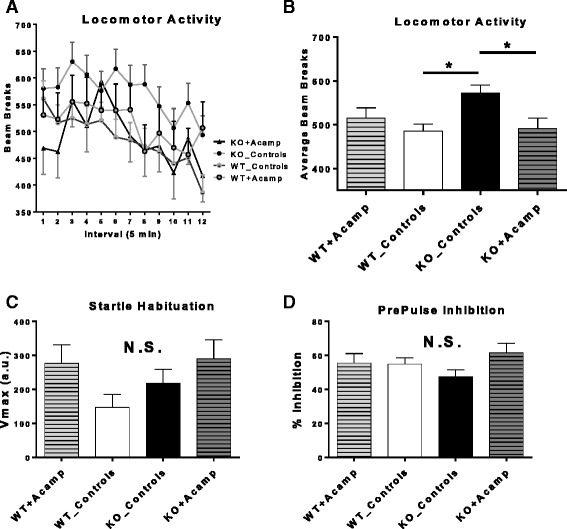
Results: Acamprosate attenuated prolonged cortical UP state duration, decreased elevated ERK1/2 activation in brain tissue, and reduced nuclear ERK1/2 activation in the dentate gyrus in KO mice. Acamprosate treatment modified behavior in anxiety and locomotor tests in Fmr1 KO mice in which control-treated KO mice were shown to deviate from control-treated WT mice. Mice treated with CaCl2 were not different from saline-treated mice in the adult behavior battery or nuclear ERK1/2 activation.
Conclusions: These data indicate that acamprosate, and not calcium, improves function reminiscent of reduced anxiety-like behavior and hyperactivity in Fmr1 KO mice and that acamprosate attenuates select electrophysiological and molecular dysregulation that may play a role in the pathophysiology of FXS. Differences between control-treated KO and WT mice were not evident in a recognition memory test or in examination of acoustic startle response/prepulse inhibition which impeded conclusions from being made about the treatment effects of acamprosate in these instances.
Keywords: Anxiety; Dendritic spine density; Electrophysiology; Extracellular signal-related kinase; Fragile X syndrome; Hippocampus; Hyperactivity; Open field; Striatum.
Figures
References
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
