

Mechanisms of Autoantibody-Induced Pathology
- PMID: 28620373
- PMCID: PMC5449453
- DOI: 10.3389/fimmu.2017.00603
Mechanisms of Autoantibody-Induced Pathology
Abstract
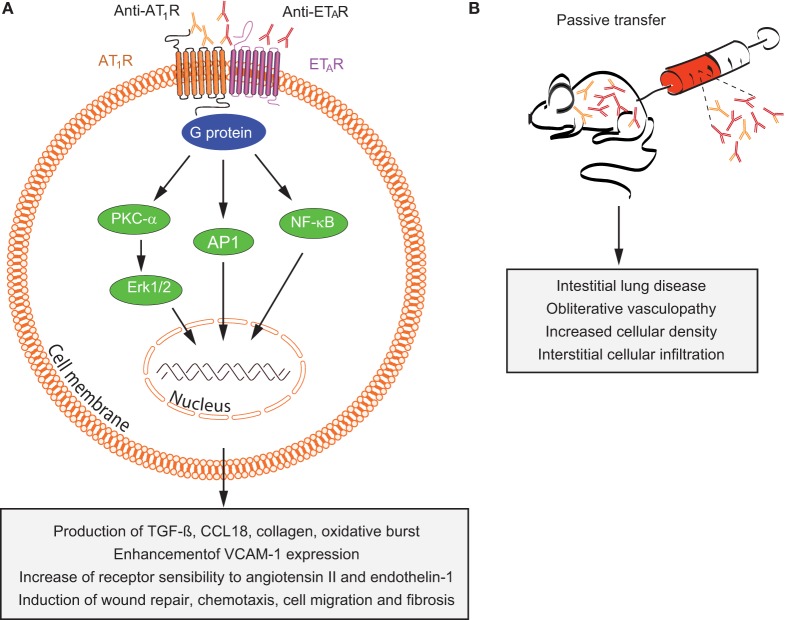
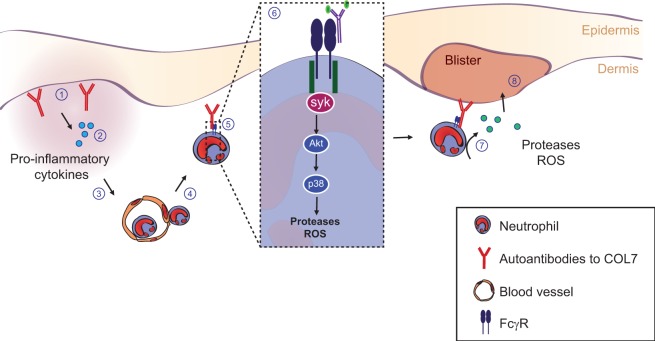
Autoantibodies are frequently observed in healthy individuals. In a minority of these individuals, they lead to manifestation of autoimmune diseases, such as rheumatoid arthritis or Graves' disease. Overall, more than 2.5% of the population is affected by autoantibody-driven autoimmune disease. Pathways leading to autoantibody-induced pathology greatly differ among different diseases, and autoantibodies directed against the same antigen, depending on the targeted epitope, can have diverse effects. To foster knowledge in autoantibody-induced pathology and to encourage development of urgently needed novel therapeutic strategies, we here categorized autoantibodies according to their effects. According to our algorithm, autoantibodies can be classified into the following categories: (1) mimic receptor stimulation, (2) blocking of neural transmission, (3) induction of altered signaling, triggering uncontrolled (4) microthrombosis, (5) cell lysis, (6) neutrophil activation, and (7) induction of inflammation. These mechanisms in relation to disease, as well as principles of autoantibody generation and detection, are reviewed herein.
Keywords: B cells; autoantibodies; autoimmunity; diagnosis; mouse models; pathogenesis; treatment.
Figures












References
-
- Vento S, Cainelli F. Autommune diseases in low and middle income countries: a neglected issue in global health. Isr Med Assoc J (2016) 18(1):54–5. - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical