

Single-cell multi-omics sequencing of mouse early embryos and embryonic stem cells
- PMID: 28621329
- PMCID: PMC5539349
- DOI: 10.1038/cr.2017.82
Single-cell multi-omics sequencing of mouse early embryos and embryonic stem cells
Abstract
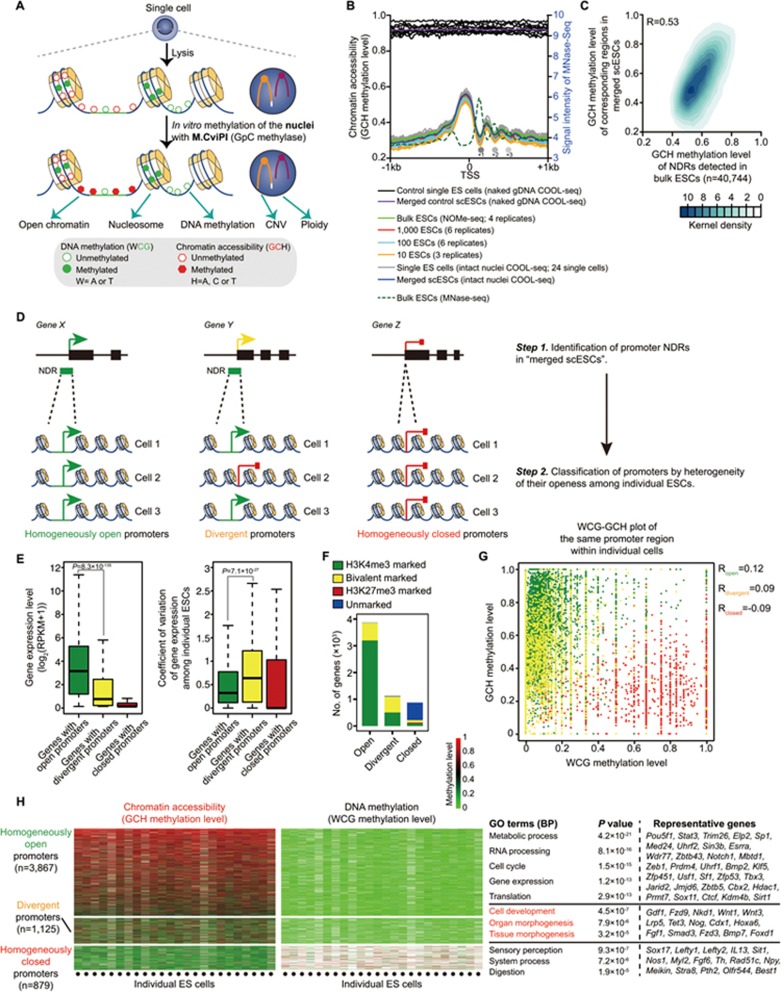
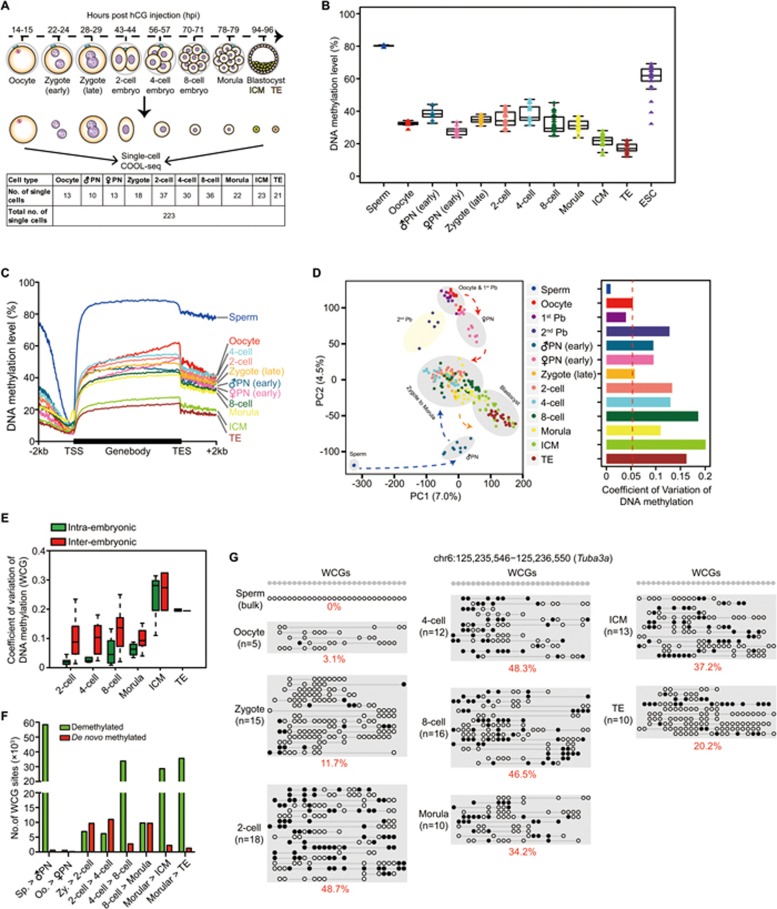
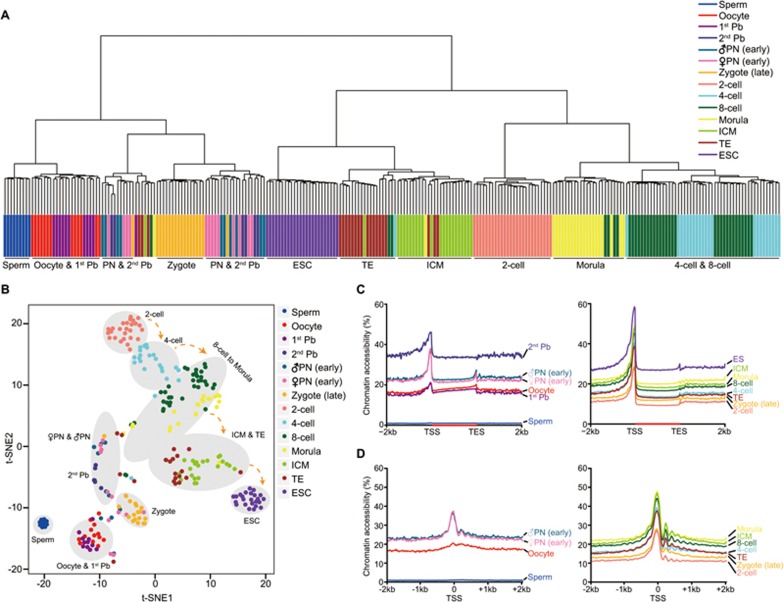
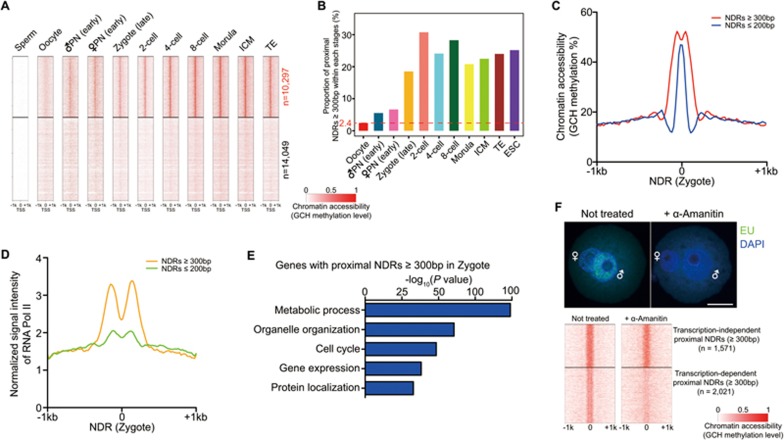
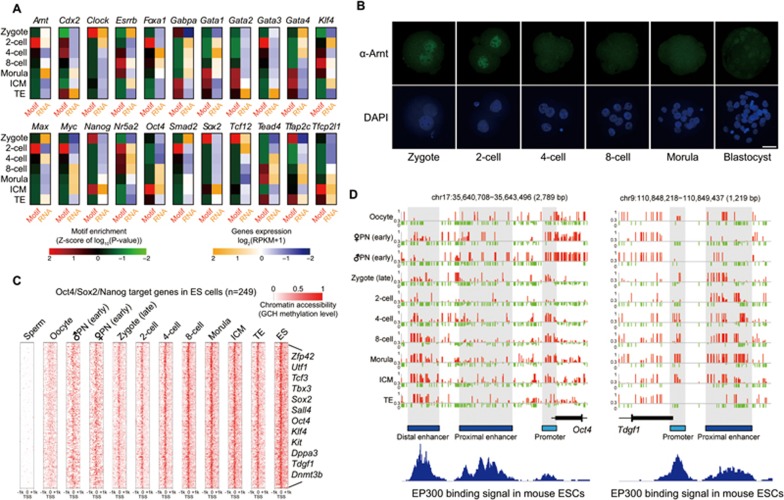
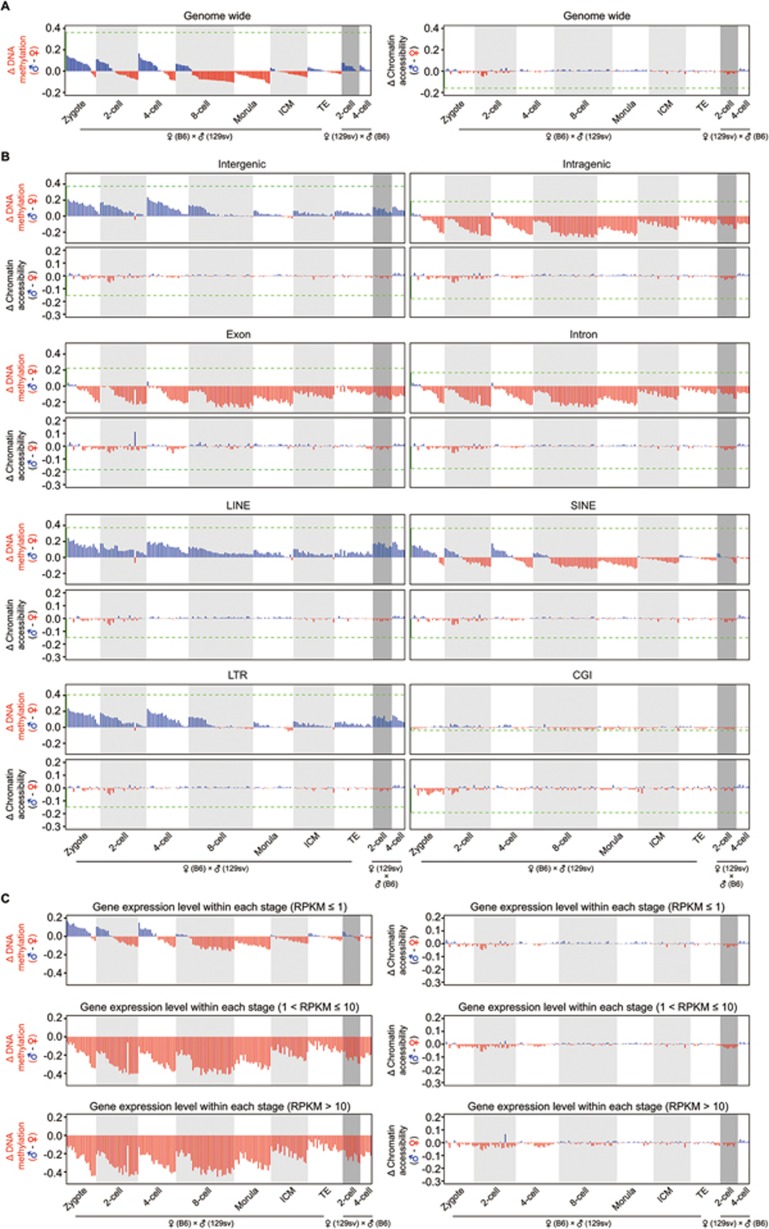
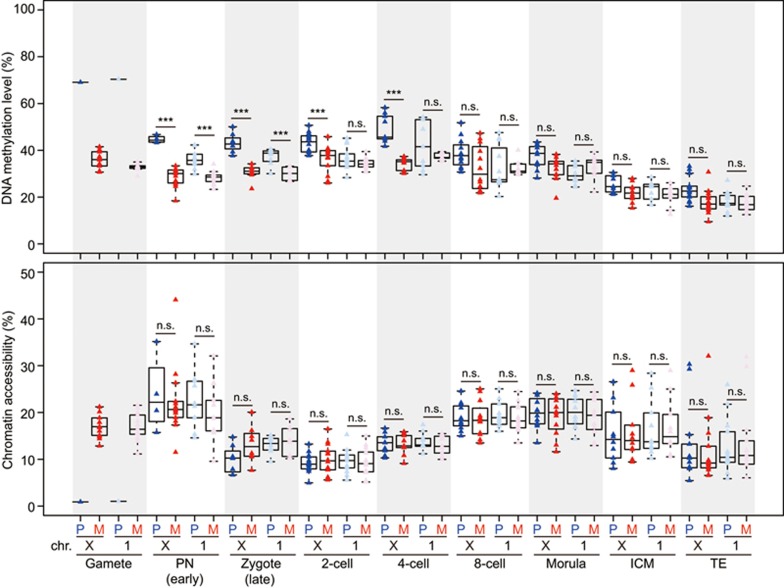
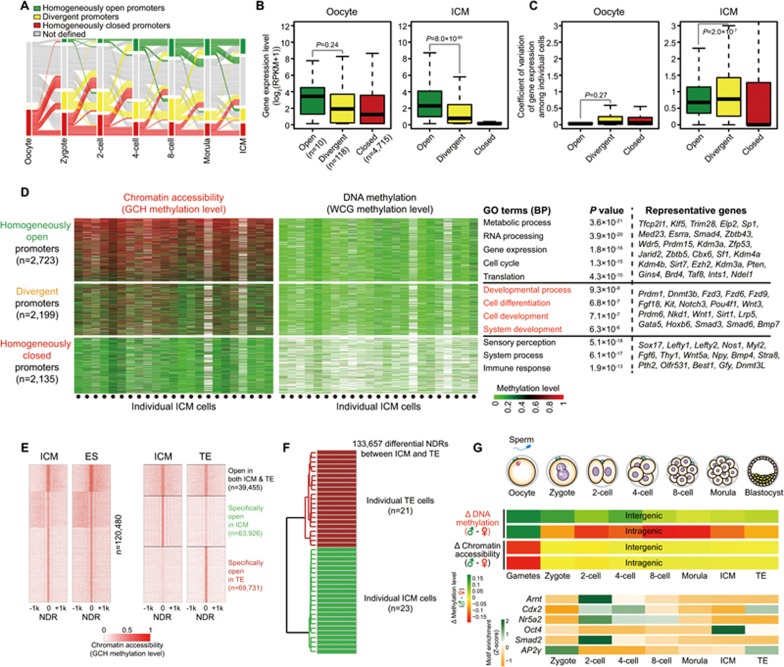
Single-cell epigenome sequencing techniques have recently been developed. However, the combination of different layers of epigenome sequencing in an individual cell has not yet been achieved. Here, we developed a single-cell multi-omics sequencing technology (single-cell COOL-seq) that can analyze the chromatin state/nucleosome positioning, DNA methylation, copy number variation and ploidy simultaneously from the same individual mammalian cell. We used this method to analyze the reprogramming of the chromatin state and DNA methylation in mouse preimplantation embryos. We found that within < 12 h of fertilization, each individual cell undergoes global genome demethylation together with the rapid and global reprogramming of both maternal and paternal genomes to a highly opened chromatin state. This was followed by decreased openness after the late zygote stage. Furthermore, from the late zygote to the 4-cell stage, the residual DNA methylation is preferentially preserved on intergenic regions of the paternal alleles and intragenic regions of maternal alleles in each individual blastomere. However, chromatin accessibility is similar between paternal and maternal alleles in each individual cell from the late zygote to the blastocyst stage. The binding motifs of several pluripotency regulators are enriched at distal nucleosome depleted regions from as early as the 2-cell stage. This indicates that the cis-regulatory elements of such target genes have been primed to an open state from the 2-cell stage onward, long before pluripotency is eventually established in the ICM of the blastocyst. Genes may be classified into homogeneously open, homogeneously closed and divergent states based on the chromatin accessibility of their promoter regions among individual cells. This can be traced to step-wise transitions during preimplantation development. Our study offers the first single-cell and parental allele-specific analysis of the genome-scale chromatin state and DNA methylation dynamics at single-base resolution in early mouse embryos and provides new insights into the heterogeneous yet highly ordered features of epigenomic reprogramming during this process.
Figures
References
-
- Gawad C, Koh W, Quake SR. Single-cell genome sequencing: current state of the science. Nat Rev Genet 2016; 17:175–188. - PubMed
-
- Huang L, Ma F, Chapman A, Lu S, Xie XS. Single-cell whole-genome amplification and sequencing: methodology and applications. Annu Rev Genomics Hum Genet 2015; 16:79–102. - PubMed
-
- Junker JP, van Oudenaarden A. Single-cell transcriptomics enters the age of mass production. Mol Cell 2015; 58:563–564. - PubMed
-
- Kolodziejczyk AA, Kim JK, Svensson V, Marioni JC, Teichmann SA. The technology and biology of single-cell RNA sequencing. Mol Cell 2015; 58:610–620. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
