

Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma
- PMID: 28622513
- PMCID: PMC5680778
- DOI: 10.1016/j.cell.2017.05.046
Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma
Abstract
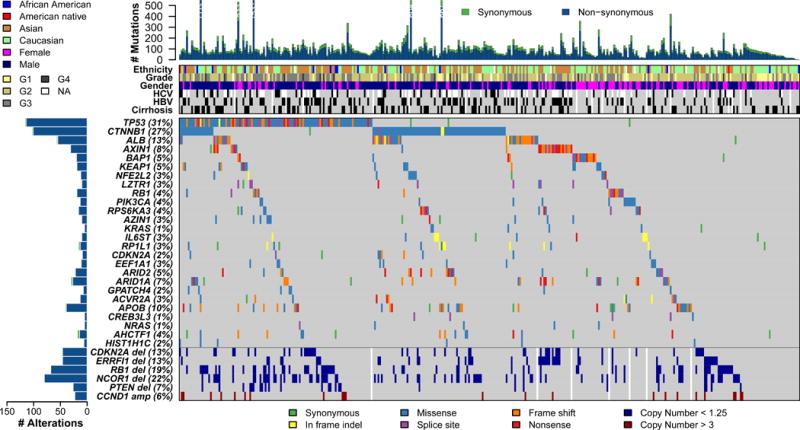
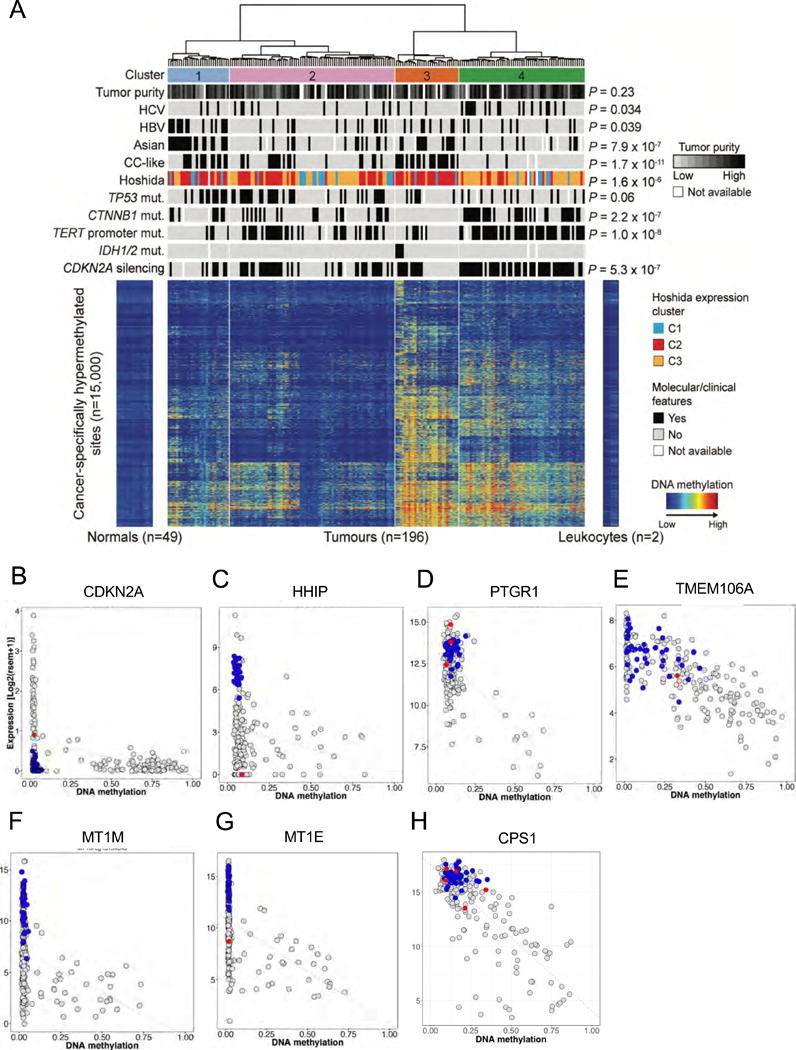
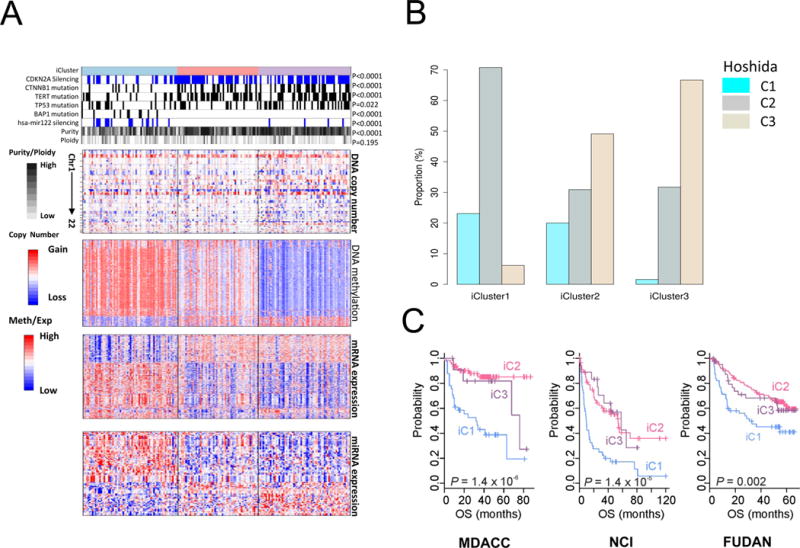
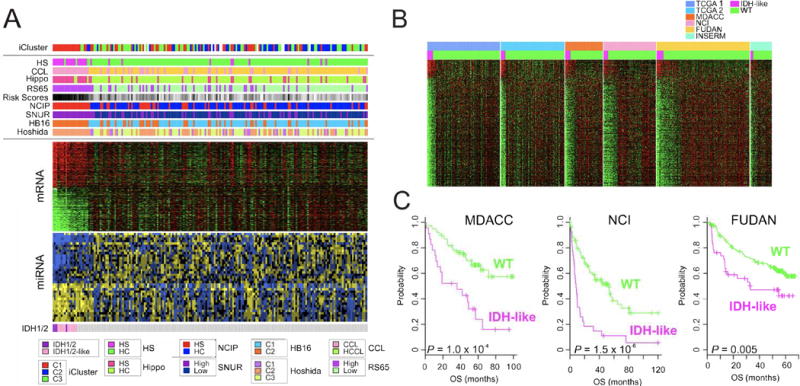
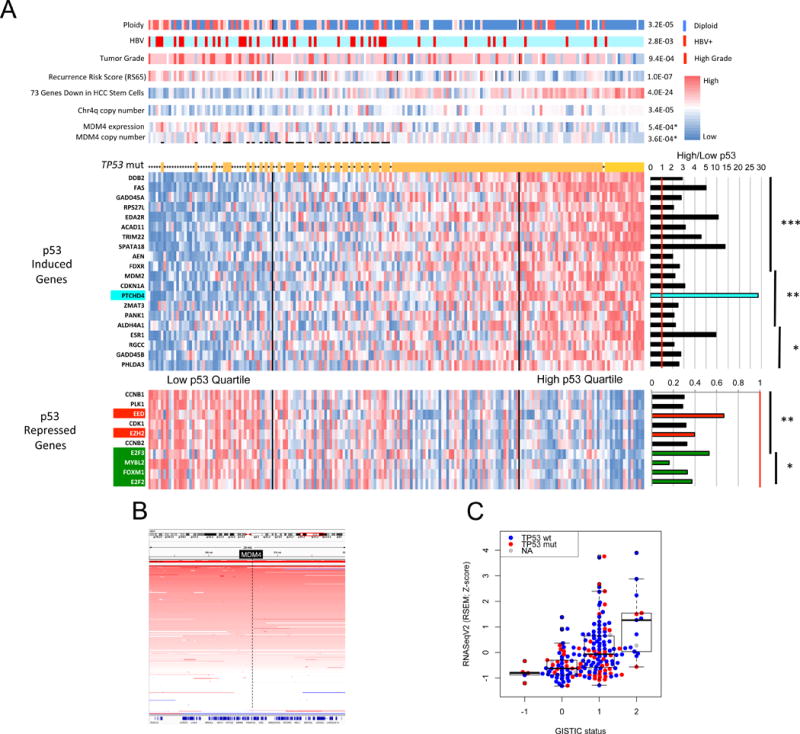
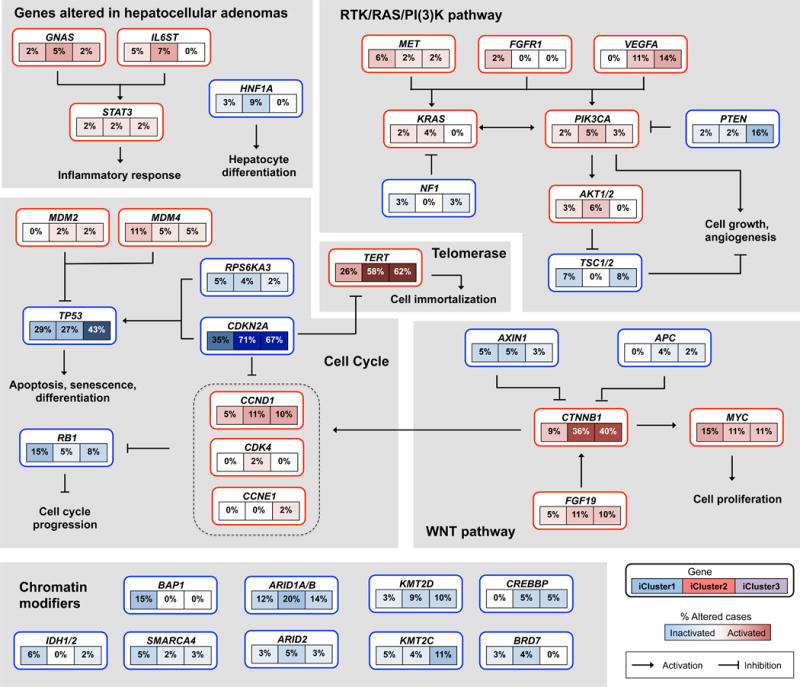
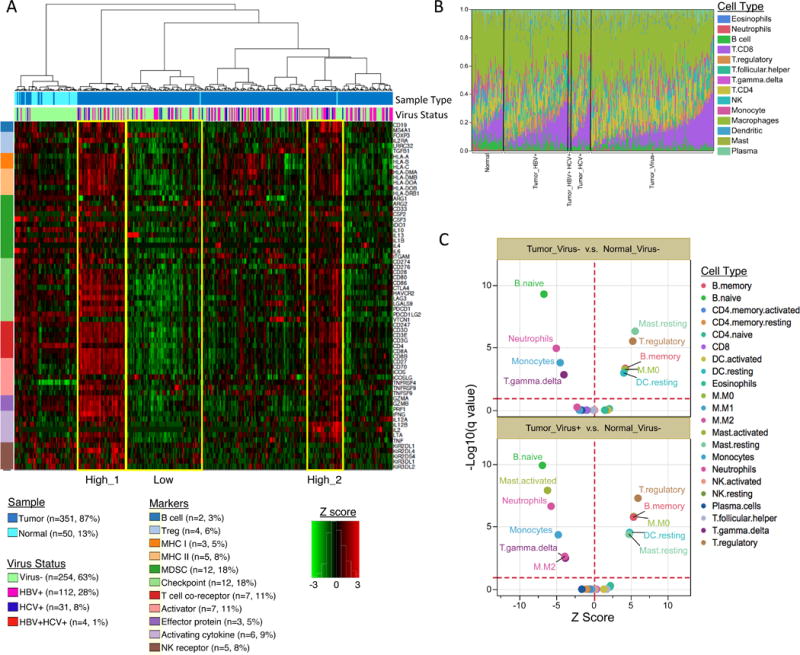
Liver cancer has the second highest worldwide cancer mortality rate and has limited therapeutic options. We analyzed 363 hepatocellular carcinoma (HCC) cases by whole-exome sequencing and DNA copy number analyses, and we analyzed 196 HCC cases by DNA methylation, RNA, miRNA, and proteomic expression also. DNA sequencing and mutation analysis identified significantly mutated genes, including LZTR1, EEF1A1, SF3B1, and SMARCA4. Significant alterations by mutation or downregulation by hypermethylation in genes likely to result in HCC metabolic reprogramming (ALB, APOB, and CPS1) were observed. Integrative molecular HCC subtyping incorporating unsupervised clustering of five data platforms identified three subtypes, one of which was associated with poorer prognosis in three HCC cohorts. Integrated analyses enabled development of a p53 target gene expression signature correlating with poor survival. Potential therapeutic targets for which inhibitors exist include WNT signaling, MDM4, MET, VEGFA, MCL1, IDH1, TERT, and immune checkpoint proteins CTLA-4, PD-1, and PD-L1.
Keywords: IDH1/2; TP53; cancer subtyping; expression profile; hepatocellular carcinoma; metabolic reprogramming; promoter hypermethylation; significantly mutated genes; sonic hedgehog signaling; stem cell phenotype.
Copyright © 2017 Elsevier Inc. All rights reserved.
Conflict of interest statement
There were no identified conflicts of interest by any of the authors.
Figures
Comment in
-
Liver cancer: Translating '-omics' results into precision medicine for hepatocellular carcinoma.Nat Rev Gastroenterol Hepatol. 2017 Oct;14(10):571-572. doi: 10.1038/nrgastro.2017.103. Epub 2017 Aug 2. Nat Rev Gastroenterol Hepatol. 2017. PMID: 28765583 No abstract available.
-
Landscape of genomic alterations in hepatocellular carcinoma: current knowledge and perspectives for targeted therapies.Hepatobiliary Surg Nutr. 2017 Dec;6(6):404-407. doi: 10.21037/hbsn.2017.10.02. Hepatobiliary Surg Nutr. 2017. PMID: 29312976 Free PMC article. No abstract available.
-
Multiplatform Genomic Roadmap of Hepatocellular Carcinoma: A Matter of Molecular Heterogeneity.Hepatology. 2018 Nov;68(5):2029-2032. doi: 10.1002/hep.29925. Epub 2018 Sep 24. Hepatology. 2018. PMID: 29637589 No abstract available.
References
-
- Ahn SM, Jang SJ, Shim JH, Kim D, Hong SM, Sung CO, Baek D, Haq F, Ansari AA, Lee SY, et al. Genomic portrait of resectable hepatocellular carcinomas: implications of RB1 and FGF19 aberrations for patient stratification. Hepatology. 2014;60:1972–1982. - PubMed
-
- Bibikova M, Barnes B, Tsan C, Ho V, Klotzle B, Le JM, Delano D, Zhang L, Schroth GP, Gunderson KL, et al. High density DNA methylation array with single CpG site resolution. Genomics. 2011;98:288–295. - PubMed
-
- Bonnal S, Vigevani L, Valcarcel J. The spliceosome as a target of novel antitumour drugs. Nat Rev Drug Discov. 2012;11:847–859. - PubMed
MeSH terms
Substances
Grants and funding
- U24 CA143882/CA/NCI NIH HHS/United States
- R01 GM066099/GM/NIGMS NIH HHS/United States
- U24 CA143866/CA/NCI NIH HHS/United States
- U54 HG003273/HG/NHGRI NIH HHS/United States
- U24 CA144025/CA/NCI NIH HHS/United States
- U24 CA143840/CA/NCI NIH HHS/United States
- U24 CA143843/CA/NCI NIH HHS/United States
- U24 CA143858/CA/NCI NIH HHS/United States
- U24 CA143848/CA/NCI NIH HHS/United States
- U24 CA210949/CA/NCI NIH HHS/United States
- U24 CA210990/CA/NCI NIH HHS/United States
- R01 GM079656/GM/NIGMS NIH HHS/United States
- P30 CA016672/CA/NCI NIH HHS/United States
- U54 HG003067/HG/NHGRI NIH HHS/United States
- U24 CA143835/CA/NCI NIH HHS/United States
- U24 CA210950/CA/NCI NIH HHS/United States
- U24 CA143845/CA/NCI NIH HHS/United States
- U24 CA143799/CA/NCI NIH HHS/United States
- I01 BX003732/BX/BLRD VA/United States
- U54 HG003079/HG/NHGRI NIH HHS/United States
- U24 CA210969/CA/NCI NIH HHS/United States
- U24 CA143883/CA/NCI NIH HHS/United States
- U24 CA210999/CA/NCI NIH HHS/United States
- U24 CA143867/CA/NCI NIH HHS/United States
- U24 CA199461/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
