

Dysbiosis in chronic periodontitis: Key microbial players and interactions with the human host
- PMID: 28623321
- PMCID: PMC5473847
- DOI: 10.1038/s41598-017-03804-8
Dysbiosis in chronic periodontitis: Key microbial players and interactions with the human host
Abstract
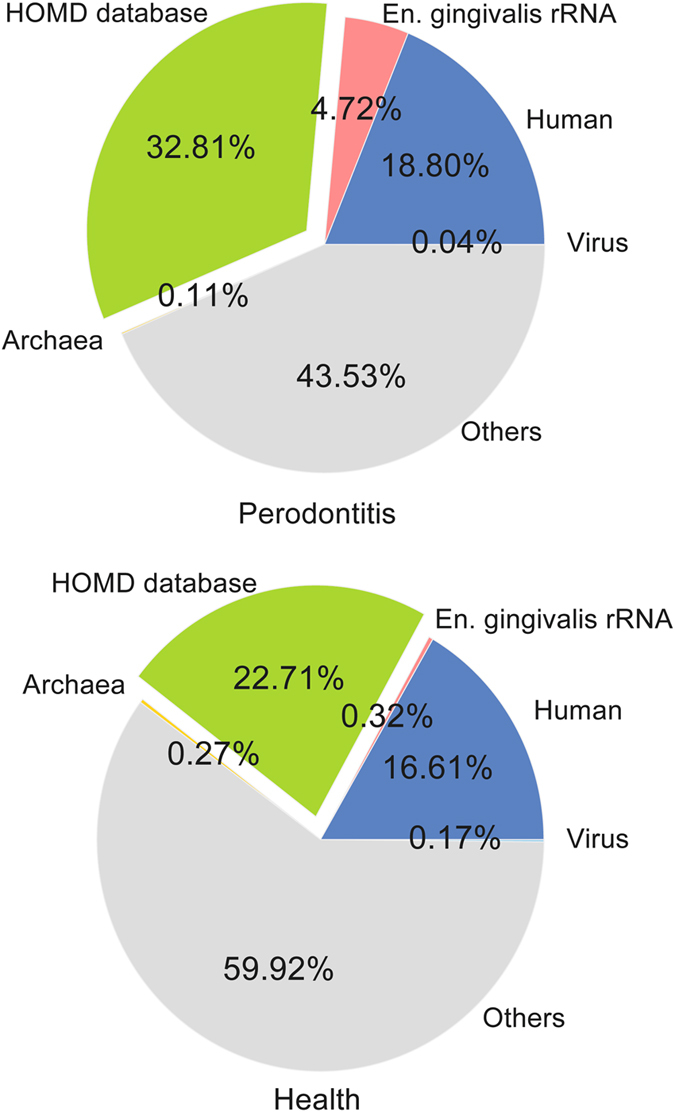
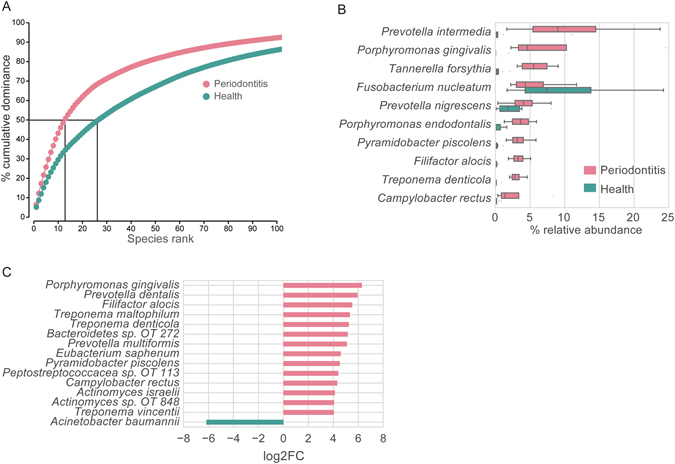
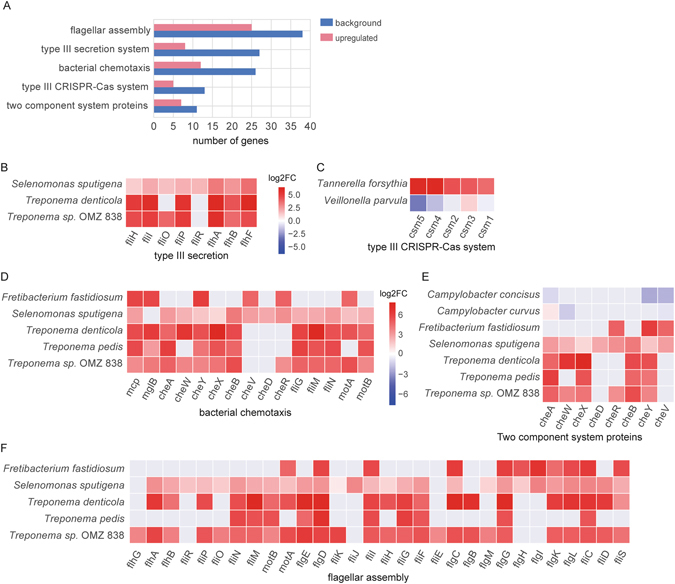
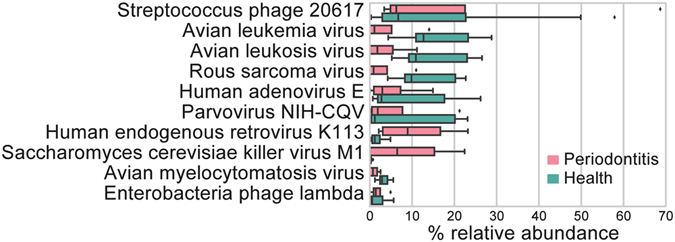
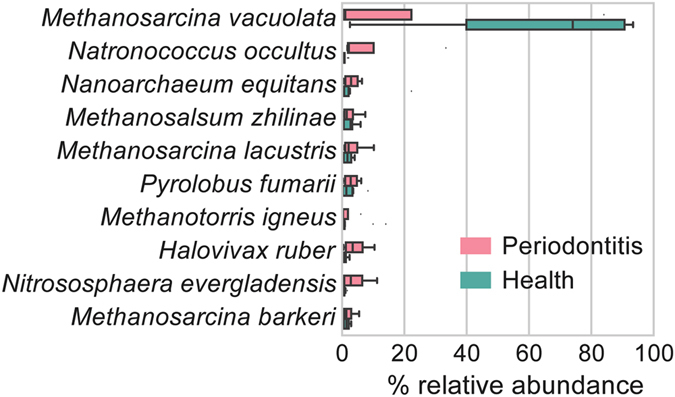
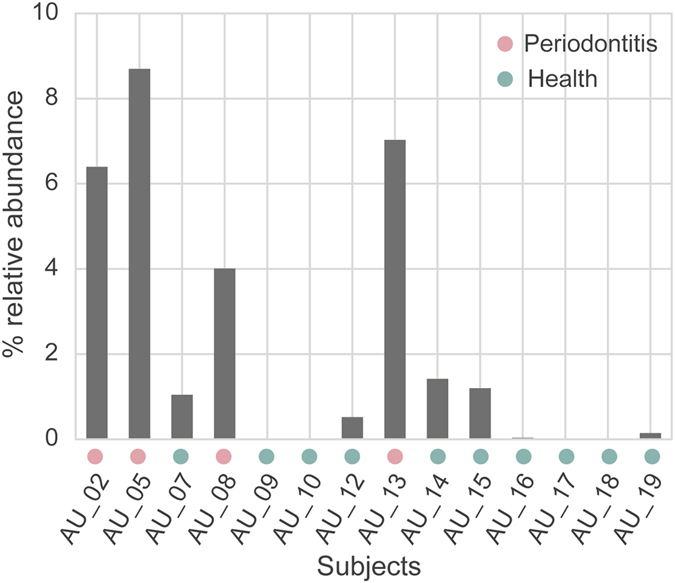
Periodontitis is an extremely prevalent disease worldwide and is driven by complex dysbiotic microbiota. Here we analyzed the transcriptional activity of the periodontal pocket microbiota from all domains of life as well as the human host in health and chronic periodontitis. Bacteria showed strong enrichment of 18 KEGG functional modules in chronic periodontitis, including bacterial chemotaxis, flagellar assembly, type III secretion system, type III CRISPR-Cas system, and two component system proteins. Upregulation of these functions was driven by the red-complex pathogens and candidate pathogens, e.g. Filifactor alocis, Prevotella intermedia, Fretibacterium fastidiosum and Selenomonas sputigena. Nine virulence factors were strongly up-regulated, among them the arginine deiminase arcA from Porphyromonas gingivalis and Mycoplasma arginini. Viruses and archaea accounted for about 0.1% and 0.22% of total putative mRNA reads, respectively, and a protozoan, Entamoeba gingivalis, was highly enriched in periodontitis. Fourteen human transcripts were enriched in periodontitis, including a gene for a ferric iron binding protein, indicating competition with the microbiota for iron, and genes associated with cancer, namely nucleolar phosphoprotein B23, ankyrin-repeat domain 30B-like protein and beta-enolase. The data provide evidence on the level of gene expression in vivo for the potentially severe impact of the dysbiotic microbiota on human health.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
