

Delineating the genetic heterogeneity of OCA in Hungarian patients
- PMID: 28629449
- PMCID: PMC5477306
- DOI: 10.1186/s40001-017-0262-0
Delineating the genetic heterogeneity of OCA in Hungarian patients
Abstract
Background: Oculocutaneous albinism (OCA) is a clinically and genetically heterogenic group of pigmentation abnormalities characterized by variable hair, skin, and ocular hypopigmentation. Six known genes and a locus on human chromosome 4q24 have been implicated in the etiology of isolated OCA forms (OCA 1-7).
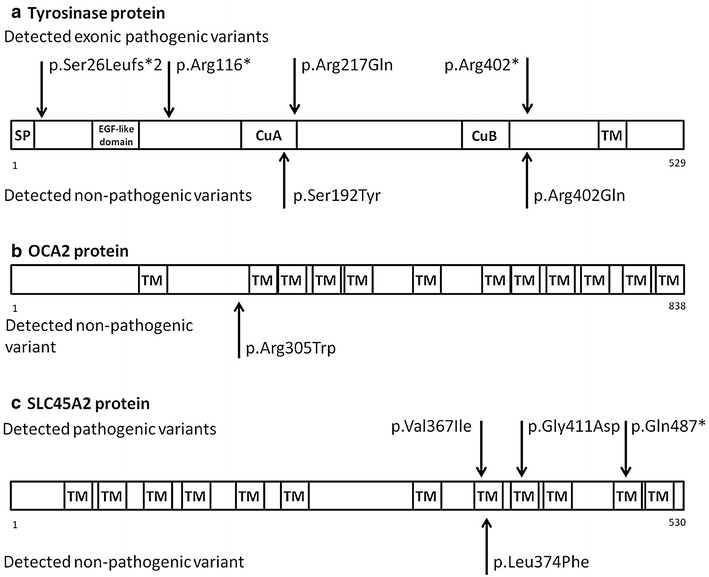
Methods: The most frequent OCA types among Caucasians are OCA1, OCA2, and OCA4. We aimed to investigate genes responsible for the development of these OCA forms in Hungarian OCA patients (n = 13). Mutation screening and polymorphism analysis were performed by direct sequencing on TYR, OCA2, SLC45A2 genes.
Results: Although the clinical features of the investigated Hungarian OCA patients were identical, the molecular genetic data suggested OCA1 subtype in eight cases and OCA4 subtype in two cases. The molecular diagnosis was not clearly identifiable in three cases. In four patients, two different heterozygous known pathogenic or predicted to be pathogenic mutations were present. Seven patients had only one pathogenic mutation, which was associated with non-pathogenic variants in six cases. In two patients no pathogenic mutation was identified.
Conclusions: Our results suggest that the concomitant screening of the non-pathogenic variants-which alone do not cause the development of OCA, but might have clinical significance in association with a pathogenic variant-is important. Our results also show significant variation in the disease spectrum compared to other populations. These data also confirm that the concomitant analysis of OCA genes is critical, providing new insights to the phenotypic diversity of OCA and expanding the mutation spectrum of OCA genes in Hungarian patients.
Keywords: Concomitant analysis; OCA2 gene; Oculocutaneous albinism; SLC45A2 gene; TYR gene.
Figures
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
