

Genetic-epigenetic interactions in cis: a major focus in the post-GWAS era
- PMID: 28629478
- PMCID: PMC5477265
- DOI: 10.1186/s13059-017-1250-y
Genetic-epigenetic interactions in cis: a major focus in the post-GWAS era
Abstract
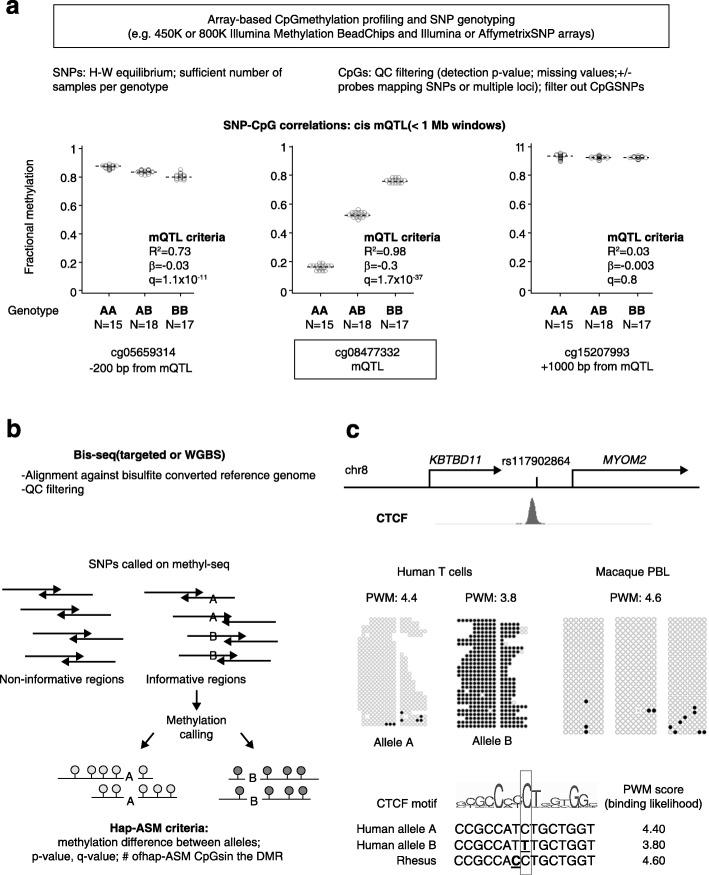
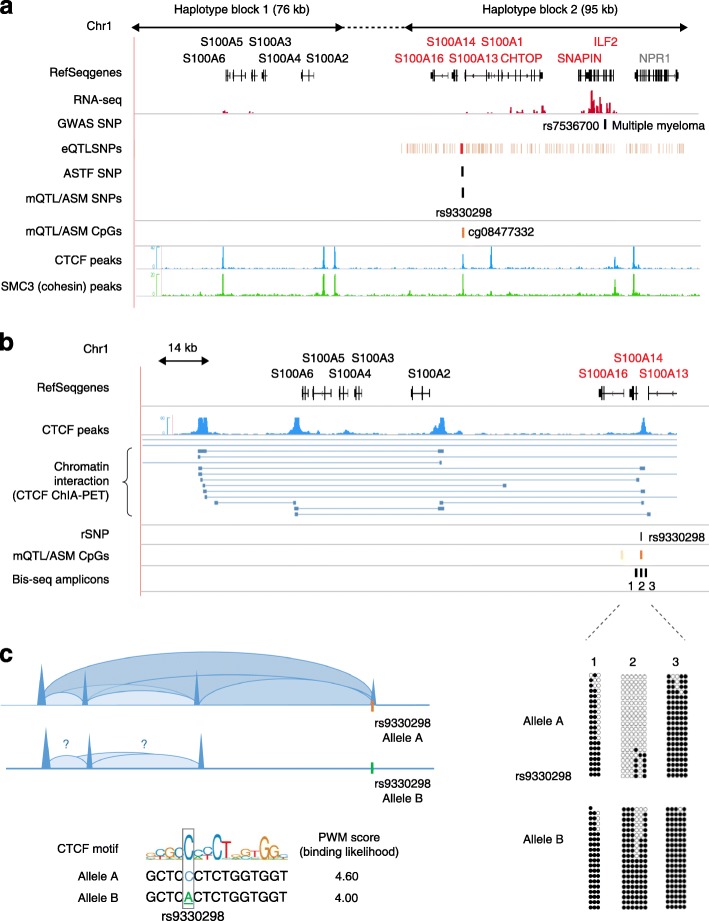
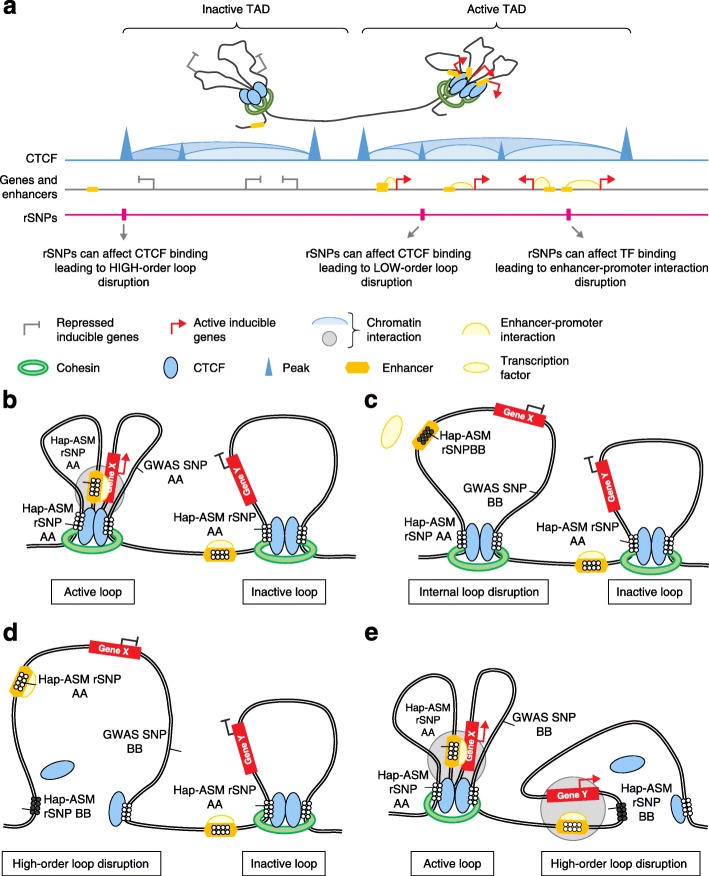
Studies on genetic-epigenetic interactions, including the mapping of methylation quantitative trait loci (mQTLs) and haplotype-dependent allele-specific DNA methylation (hap-ASM), have become a major focus in the post-genome-wide-association-study (GWAS) era. Such maps can nominate regulatory sequence variants that underlie GWAS signals for common diseases, ranging from neuropsychiatric disorders to cancers. Conversely, mQTLs need to be filtered out when searching for non-genetic effects in epigenome-wide association studies (EWAS). Sequence variants in CCCTC-binding factor (CTCF) and transcription factor binding sites have been mechanistically linked to mQTLs and hap-ASM. Identifying these sites can point to disease-associated transcriptional pathways, with implications for targeted treatment and prevention.
Figures
References
-
- Korf BR. Integration of genomics into medical practice. Discov Med. 2013;16:241–8. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
