

Virus-induced inflammasome activation is suppressed by prostaglandin D2/DP1 signaling
- PMID: 28630327
- PMCID: PMC5502630
- DOI: 10.1073/pnas.1704099114
Virus-induced inflammasome activation is suppressed by prostaglandin D2/DP1 signaling
Abstract
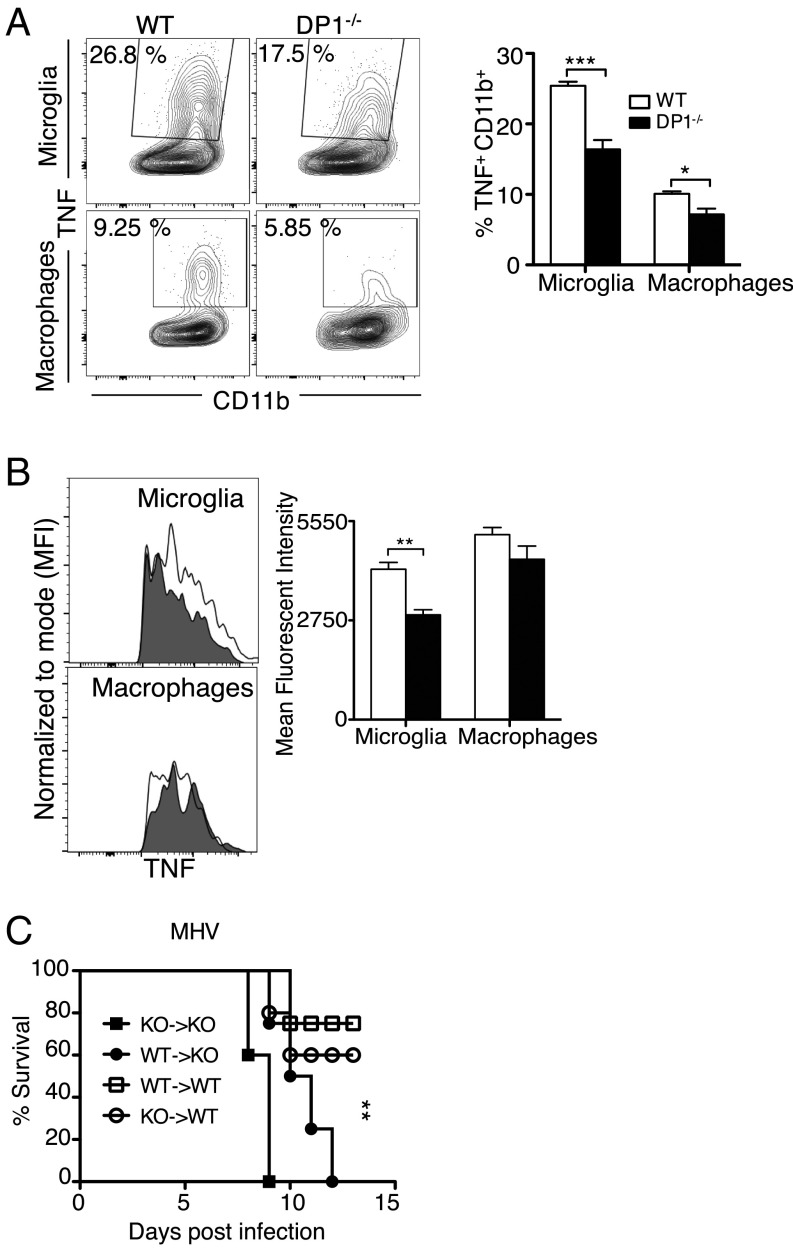
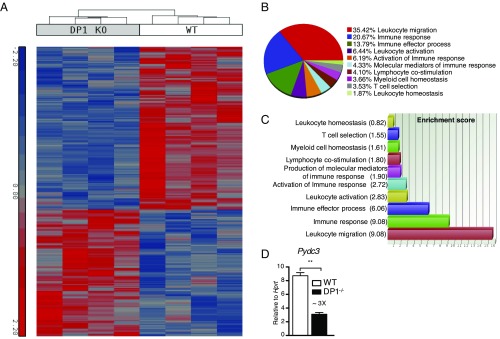
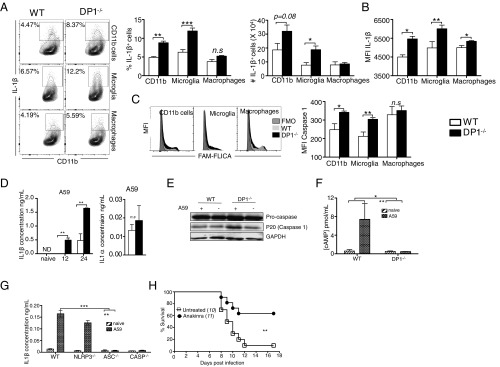
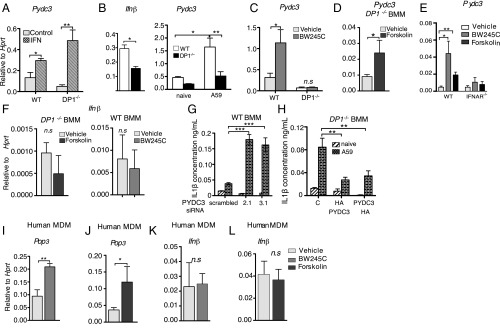
Prostaglandin D2 (PGD2), an eicosanoid with both pro- and anti-inflammatory properties, is the most abundantly expressed prostaglandin in the brain. Here we show that PGD2 signaling through the D-prostanoid receptor 1 (DP1) receptor is necessary for optimal microglia/macrophage activation and IFN expression after infection with a neurotropic coronavirus. Genome-wide expression analyses indicated that PGD2/DP1 signaling is required for up-regulation of a putative inflammasome inhibitor, PYDC3, in CD11b+ cells in the CNS of infected mice. Our results also demonstrated that, in addition to PGD2/DP1 signaling, type 1 IFN (IFN-I) signaling is required for PYDC3 expression. In the absence of Pydc3 up-regulation, IL-1β expression and, subsequently, mortality were increased in infected DP1-/- mice. Notably, survival was enhanced by IL1 receptor blockade, indicating that the effects of the absence of DP1 signaling on clinical outcomes were mediated, at least in part, by inflammasomes. Using bone marrow-derived macrophages in vitro, we confirmed that PYDC3 expression is dependent upon DP1 signaling and that IFN priming is critical for PYDC3 up-regulation. In addition, Pydc3 silencing or overexpression augmented or diminished IL-1β secretion, respectively. Furthermore, DP1 signaling in human macrophages also resulted in the up-regulation of a putative functional analog, POP3, suggesting that PGD2 similarly modulates inflammasomes in human cells. These findings demonstrate a previously undescribed role for prostaglandin signaling in preventing excessive inflammasome activation and, together with previously published results, suggest that eicosanoids and inflammasomes are reciprocally regulated.
Keywords: coronavirus; encephalitis; inflammasomes; prostaglandin D2; pyrin domain-only protein.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
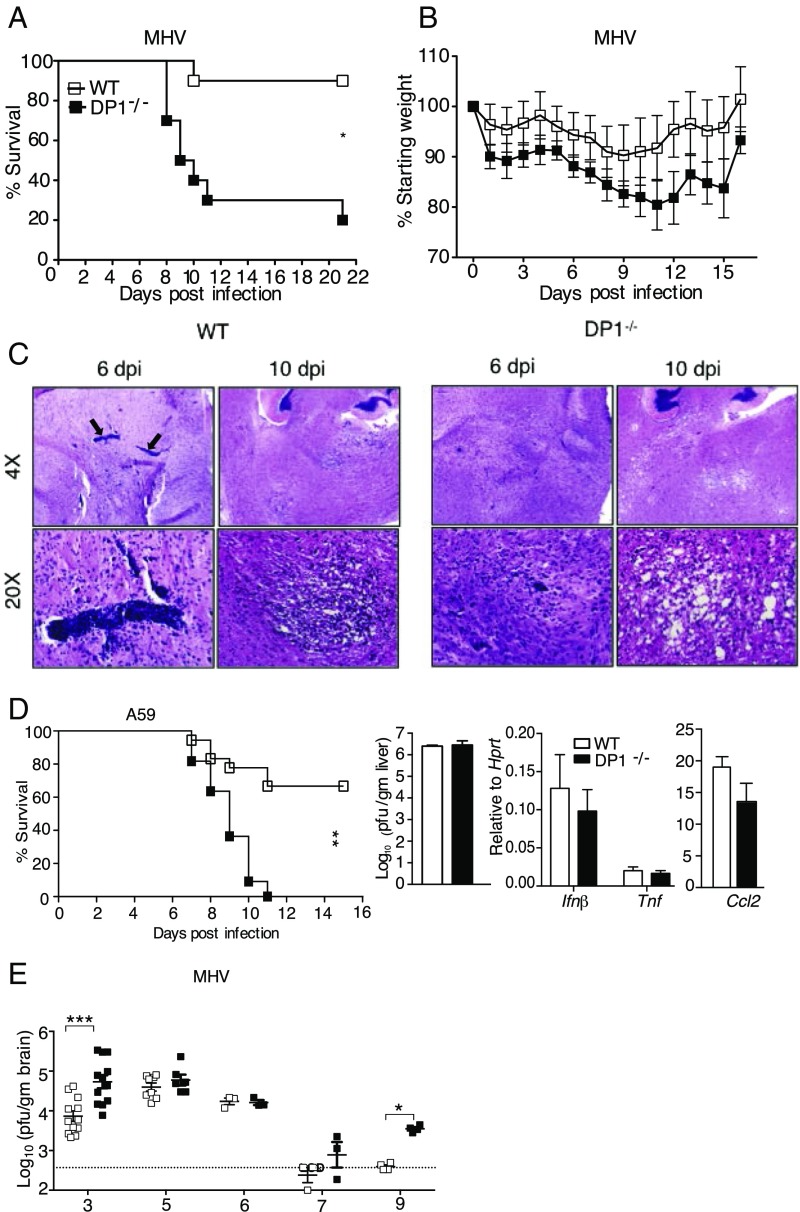
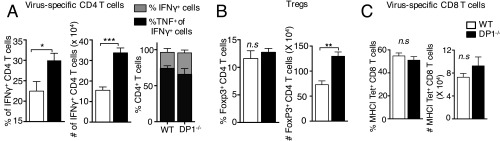
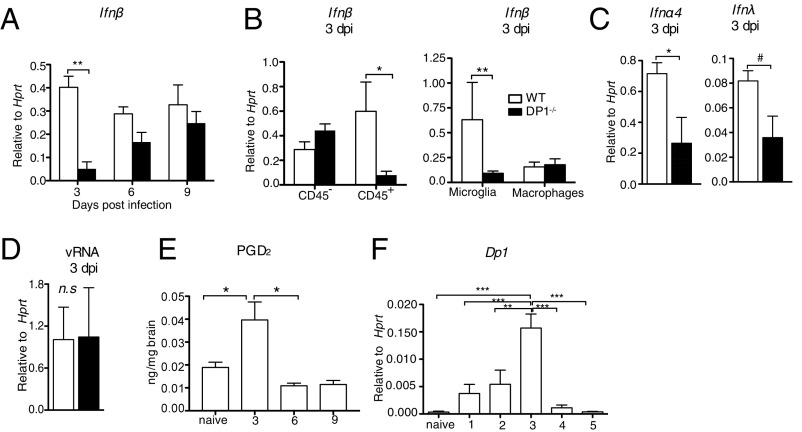
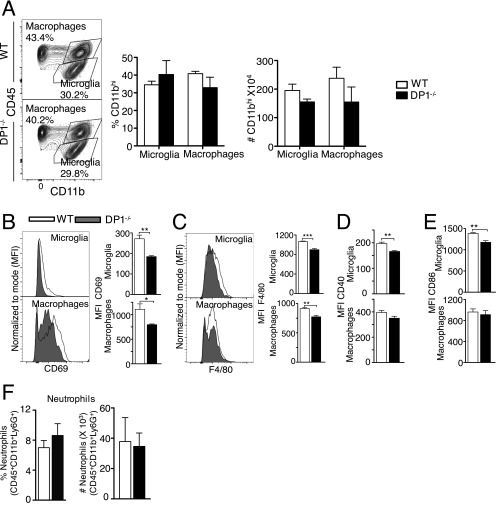
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
