

Global patterns in coronavirus diversity
- PMID: 28630747
- PMCID: PMC5467638
- DOI: 10.1093/ve/vex012
Global patterns in coronavirus diversity
Abstract
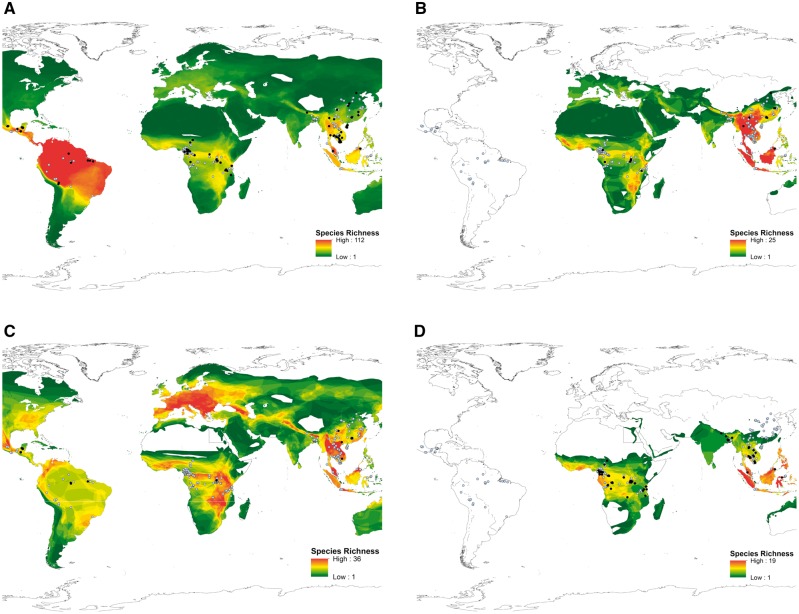
Since the emergence of Severe Acute Respiratory Syndrome Coronavirus (SARS-CoV) and Middle East Respiratory Syndrom Coronavirus (MERS-CoV) it has become increasingly clear that bats are important reservoirs of CoVs. Despite this, only 6% of all CoV sequences in GenBank are from bats. The remaining 94% largely consist of known pathogens of public health or agricultural significance, indicating that current research effort is heavily biased towards describing known diseases rather than the 'pre-emergent' diversity in bats. Our study addresses this critical gap, and focuses on resource poor countries where the risk of zoonotic emergence is believed to be highest. We surveyed the diversity of CoVs in multiple host taxa from twenty countries to explore the factors driving viral diversity at a global scale. We identified sequences representing 100 discrete phylogenetic clusters, ninety-one of which were found in bats, and used ecological and epidemiologic analyses to show that patterns of CoV diversity correlate with those of bat diversity. This cements bats as the major evolutionary reservoirs and ecological drivers of CoV diversity. Co-phylogenetic reconciliation analysis was also used to show that host switching has contributed to CoV evolution, and a preliminary analysis suggests that regional variation exists in the dynamics of this process. Overall our study represents a model for exploring global viral diversity and advances our fundamental understanding of CoV biodiversity and the potential risk factors associated with zoonotic emergence.
Keywords: bat; coronavirus; evolution; viral ecology.
Figures
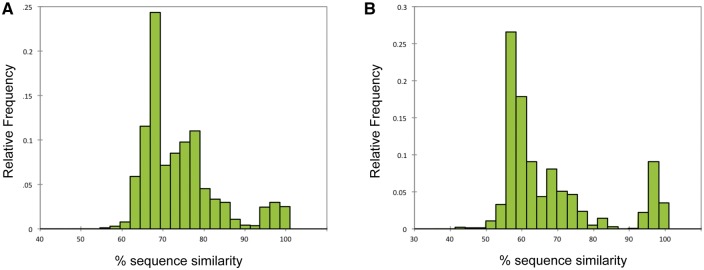
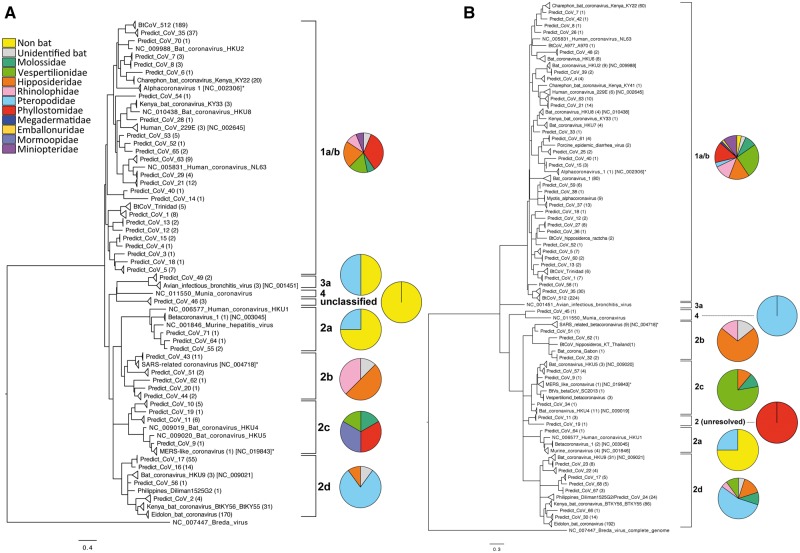

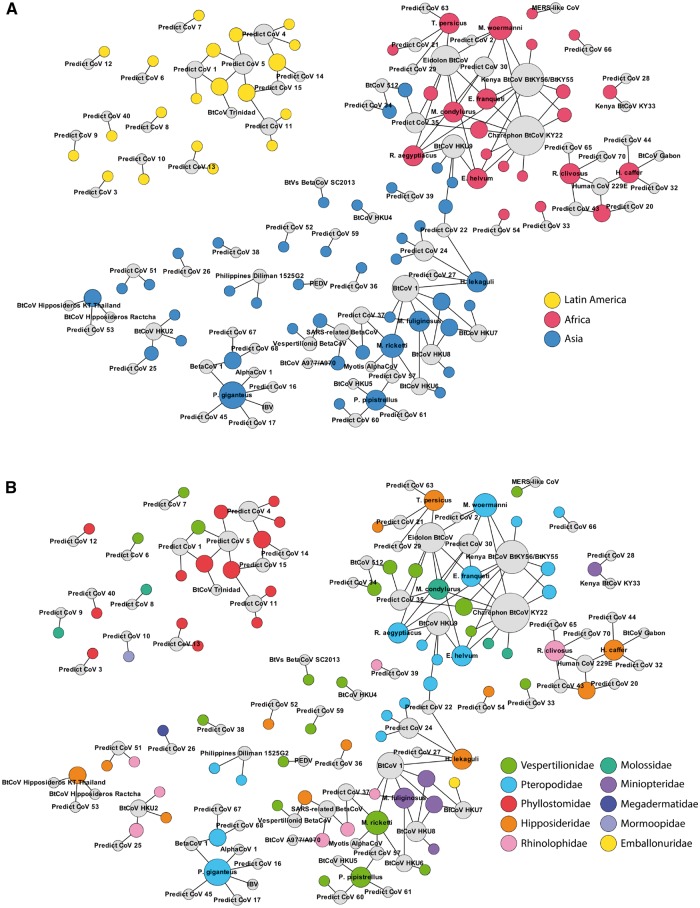

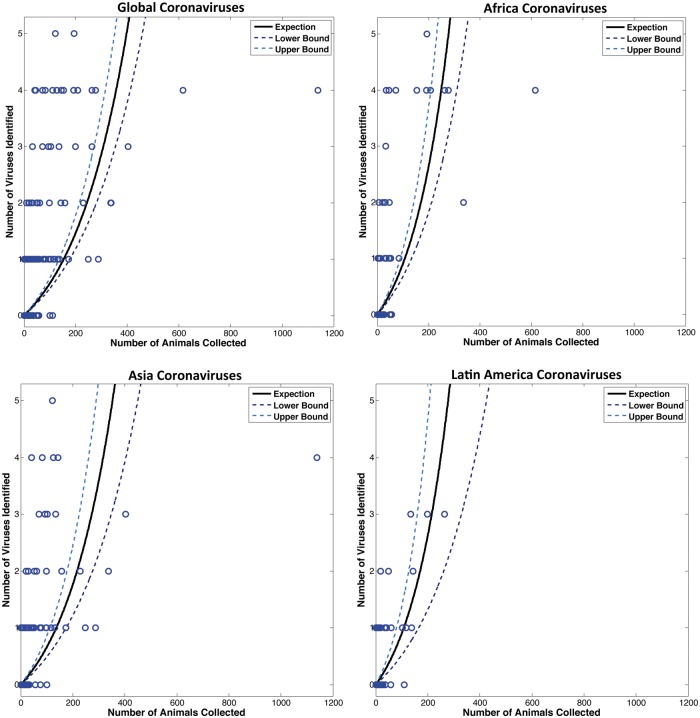
Comment in
-
Bats are global reservoir for deadly coronaviruses.Nature. 2017 Jun 12;546(7658):340. doi: 10.1038/nature.2017.22137. Nature. 2017. PMID: 28617480 No abstract available.
References
-
- Azhar E. I. et al. (2014) ‘Evidence for camel-to-human transmission of MERS coronavirus’, The New England Journal of Medicine, 370: 2499–505. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
