

The multiple pathways to autoimmunity
- PMID: 28632714
- PMCID: PMC5791156
- DOI: 10.1038/ni.3731
The multiple pathways to autoimmunity
Abstract
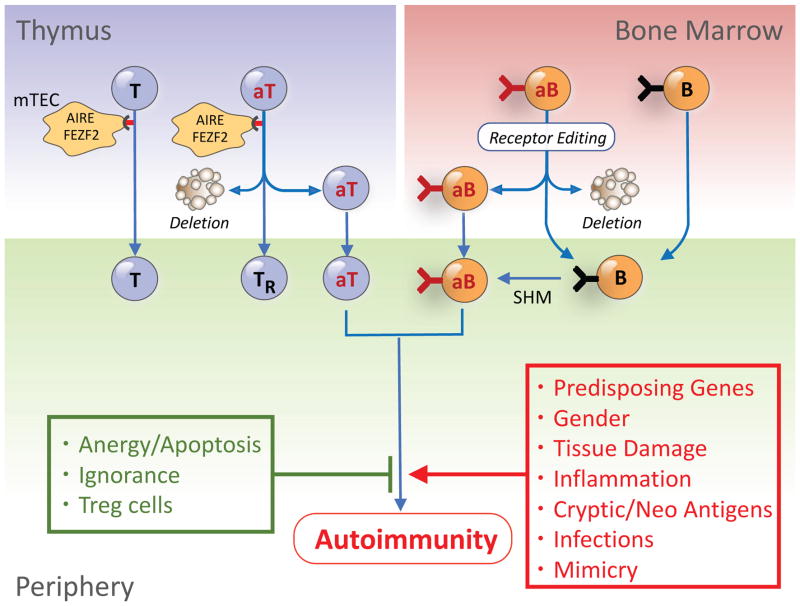
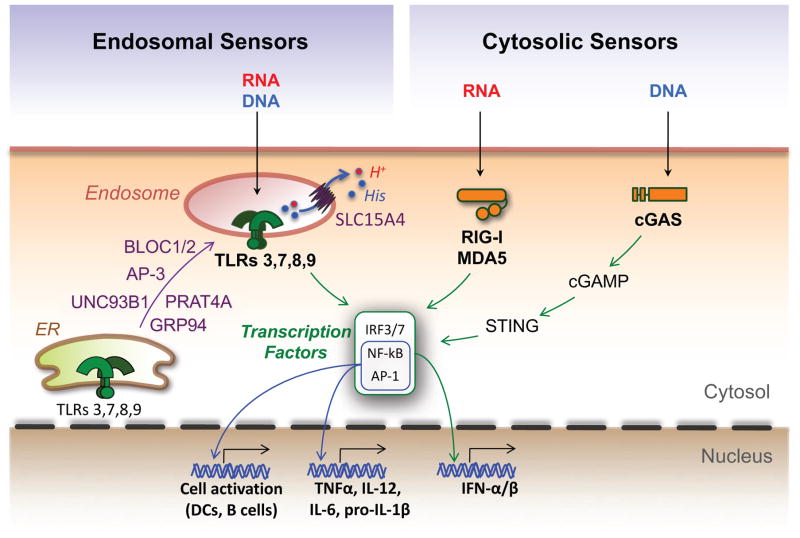
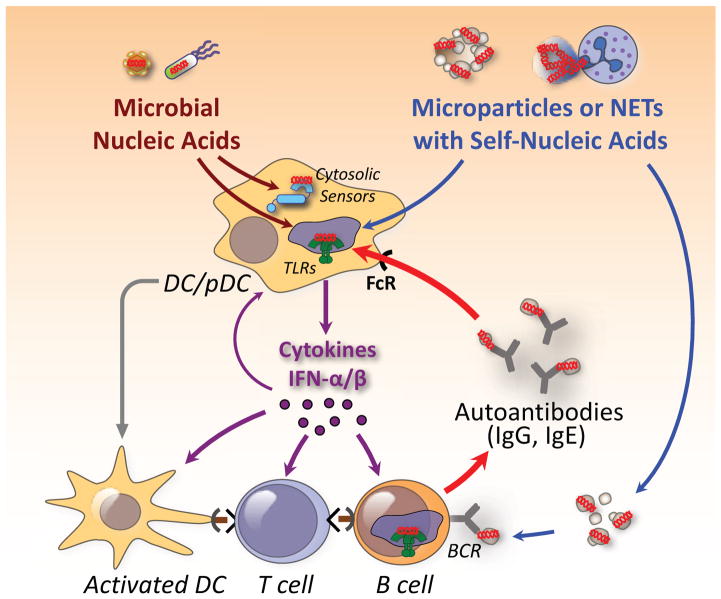
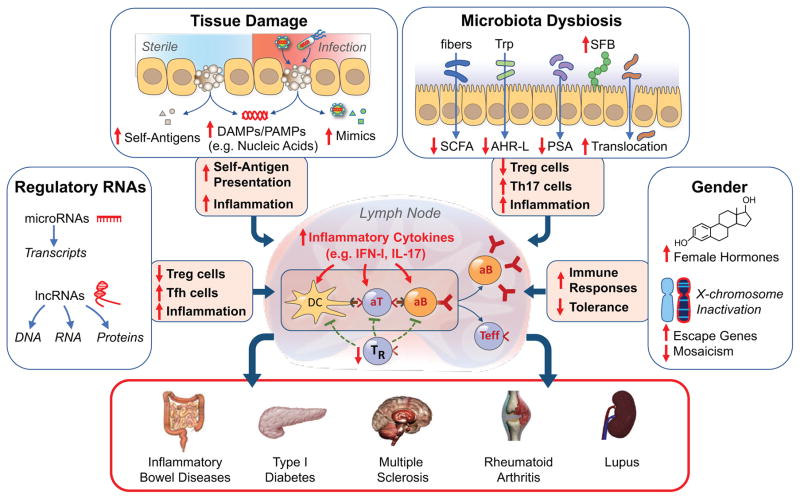
Efforts to understand autoimmunity have been pursued relentlessly for several decades. It has become apparent that the immune system evolved multiple mechanisms for controlling self-reactivity, and defects in one or more of these mechanisms can lead to a breakdown of tolerance. Among the multitude of lesions associated with disease, the most common seem to affect peripheral tolerance rather than central tolerance. The initial trigger for both systemic autoimmune disorders and organ-specific autoimmune disorders probably involves the recognition of self or foreign molecules, especially nucleic acids, by innate sensors. Such recognition, in turn, triggers inflammatory responses and the engagement of previously quiescent autoreactive T cells and B cells. Here we summarize the most prominent autoimmune pathways and identify key issues that require resolution for full understanding of pathogenic autoimmunity.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
References
-
- Kono DH, Theofilopoulos AN. Kelley and Firestein’s Textbook of Rheumatology. 10. Elsevier; Philadelphia: 2017. Autoimmunity; pp. 301–317.
-
- Bouneaud C, Kourilsky P, Bousso P. Impact of negative selection on the T cell repertoire reactive to a self-peptide: a large fraction of T cell clones escapes clonal deletion. Immunity. 2000;13:829–840. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
