

Apoptosis and Compensatory Proliferation Signaling Are Coupled by CrkI-Containing Microvesicles
- PMID: 28633020
- PMCID: PMC5533184
- DOI: 10.1016/j.devcel.2017.05.014
Apoptosis and Compensatory Proliferation Signaling Are Coupled by CrkI-Containing Microvesicles
Abstract
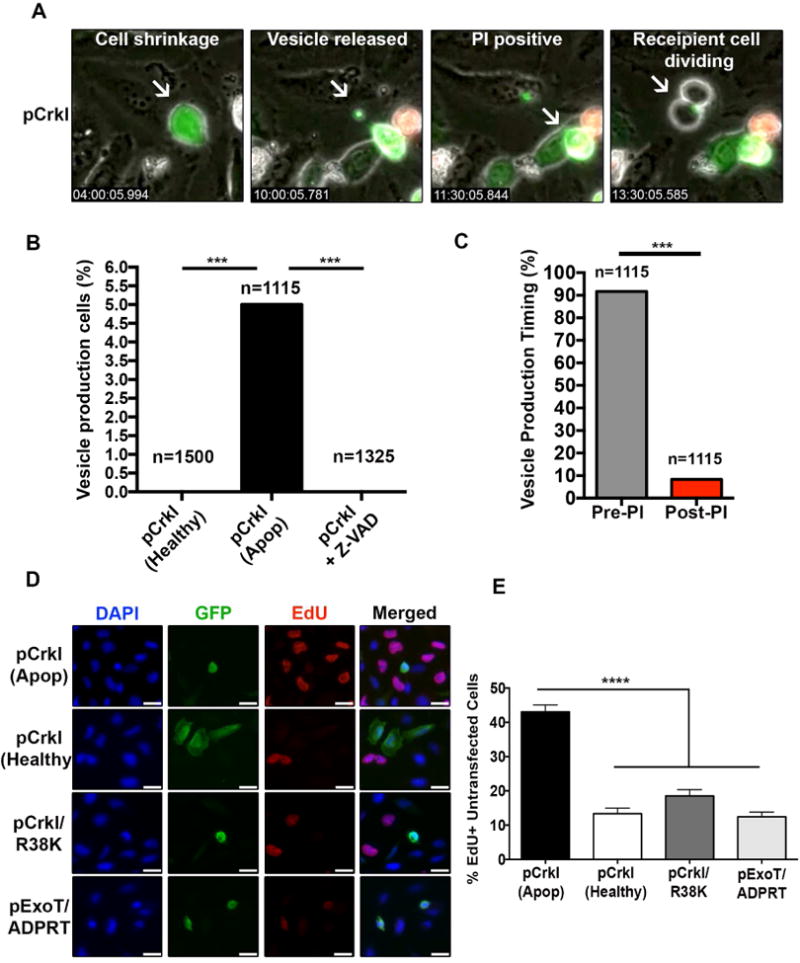
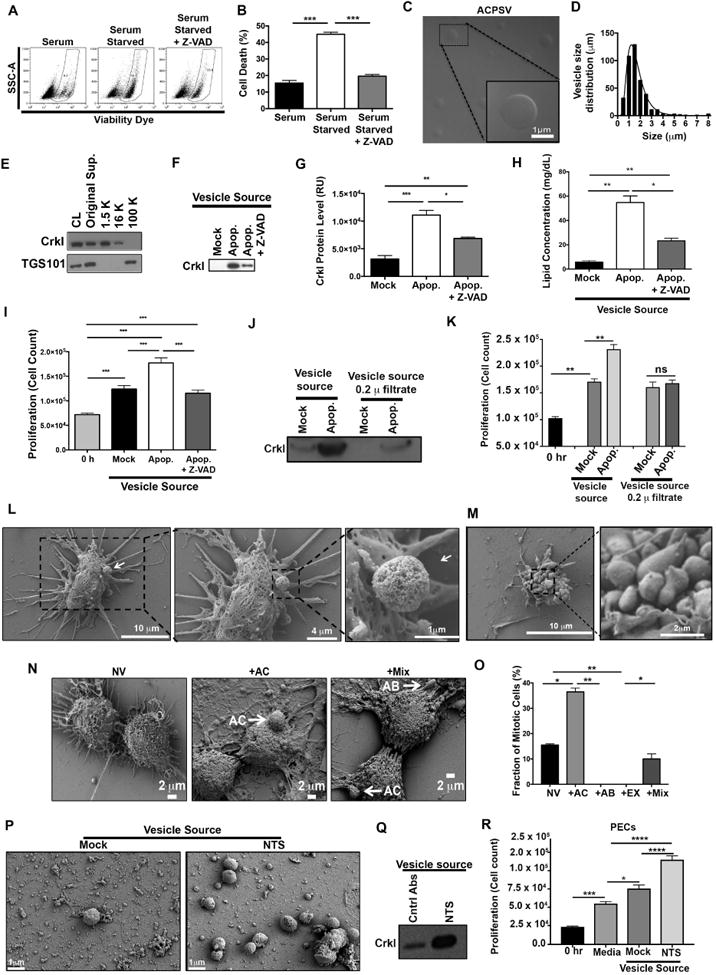
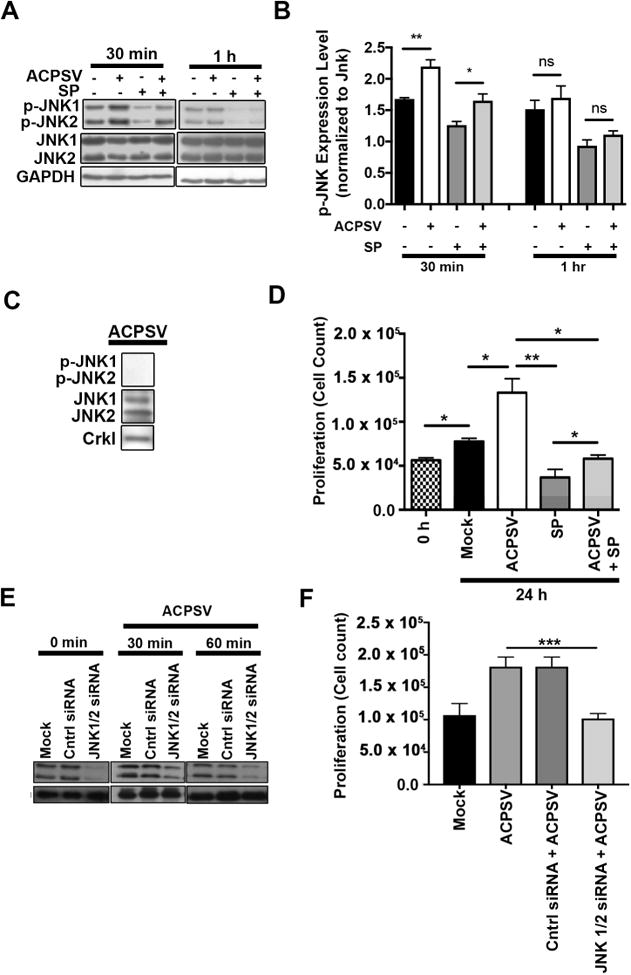
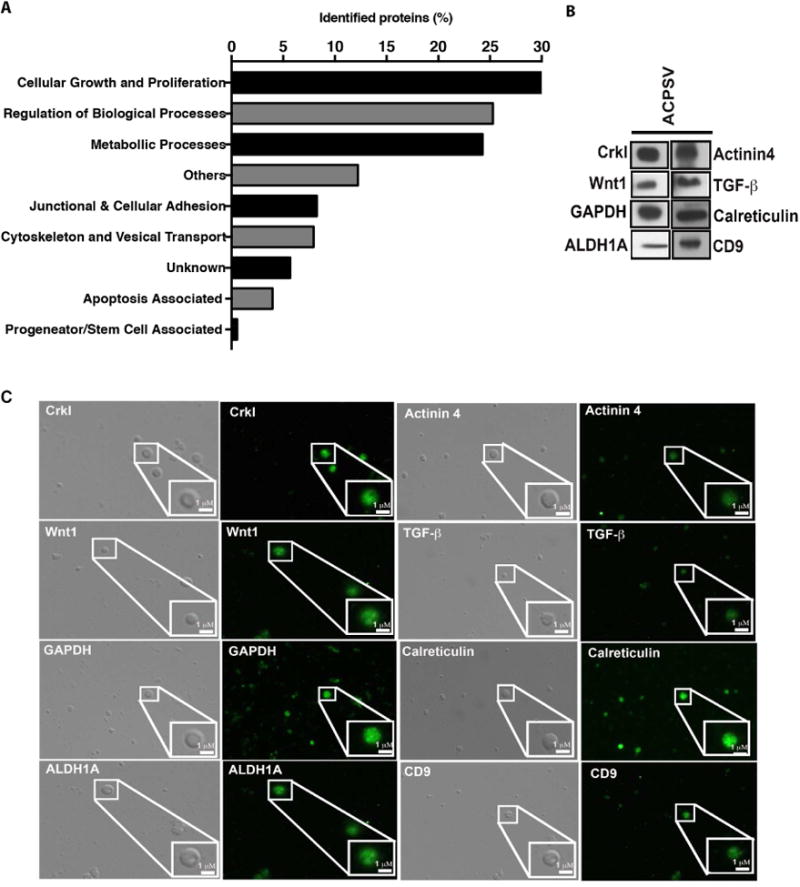
Apoptosis has been implicated in compensatory proliferation signaling (CPS), whereby dying cells induce proliferation in neighboring cells as a means to restore homeostasis. The nature of signaling between apoptotic cells and their neighboring cells remains largely unknown. Here we show that a fraction of apoptotic cells produce and release CrkI-containing microvesicles (distinct from exosomes and apoptotic bodies), which induce proliferation in neighboring cells upon contact. We provide visual evidence of CPS by videomicroscopy. We show that purified vesicles in vitro and in vivo are sufficient to stimulate proliferation in other cells. Our data demonstrate that CrkI inactivation by ExoT bacterial toxin or by mutagenesis blocks vesicle formation in apoptotic cells and inhibits CPS, thus uncoupling apoptosis from CPS. We further show that c-Jun amino-terminal kinase (JNK) plays a pivotal role in mediating vesicle-induced CPS in recipient cells. CPS could have important ramifications in diseases that involve apoptotic cell death.
Keywords: ACPS; ACPSV; CT10 regulator of kinase I; CrkI; ExoT; Exotoxin T; JNK; NTS; Pseudomonas aeruginosa; apoptosis; apoptotic compensatory proliferation signaling; apoptotic compensatory proliferation signaling vesicle; c-Jun amino-terminal kinase; glomerulonephritis; nephritis; nephrotoxic serum.
Copyright © 2017 Elsevier Inc. All rights reserved.
Conflict of interest statement
Figures
References
-
- Bergantinos C, Corominas M, Serras F. Cell death-induced regeneration in wing imaginal discs requires JNK signalling. Development. 2010;137:1169–1179. - PubMed
-
- Bouchard V, Harnois C, Demers MJ, Thibodeau S, Laquerre V, Gauthier R, Vezina A, Noel D, Fujita N, Tsuruo T, et al. B1 integrin/Fak/Src signaling in intestinal epithelial crypt cell survival: integration of complex regulatory mechanisms. Apoptosis: an international journal on programmed cell death. 2008;13:531–542. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
