

Emerging mechanisms of aminoacyl-tRNA synthetase mutations in recessive and dominant human disease
- PMID: 28633377
- PMCID: PMC5886470
- DOI: 10.1093/hmg/ddx231
Emerging mechanisms of aminoacyl-tRNA synthetase mutations in recessive and dominant human disease
Abstract
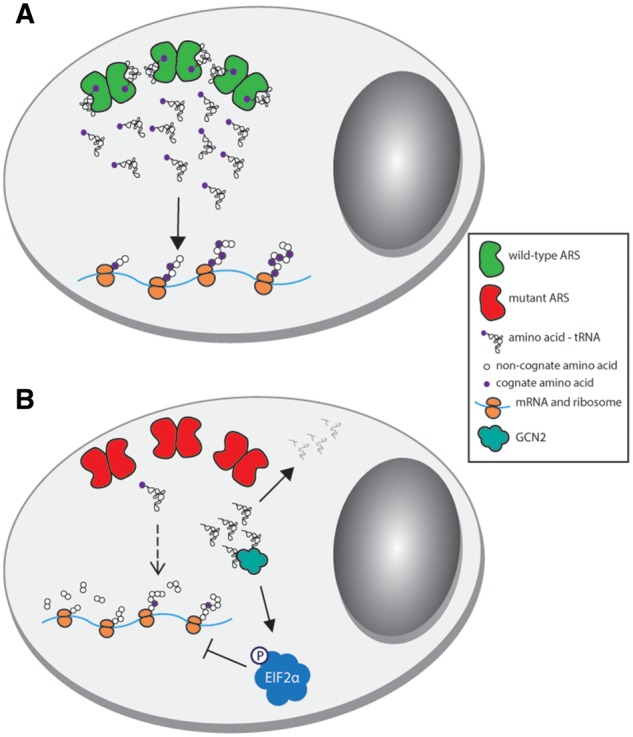
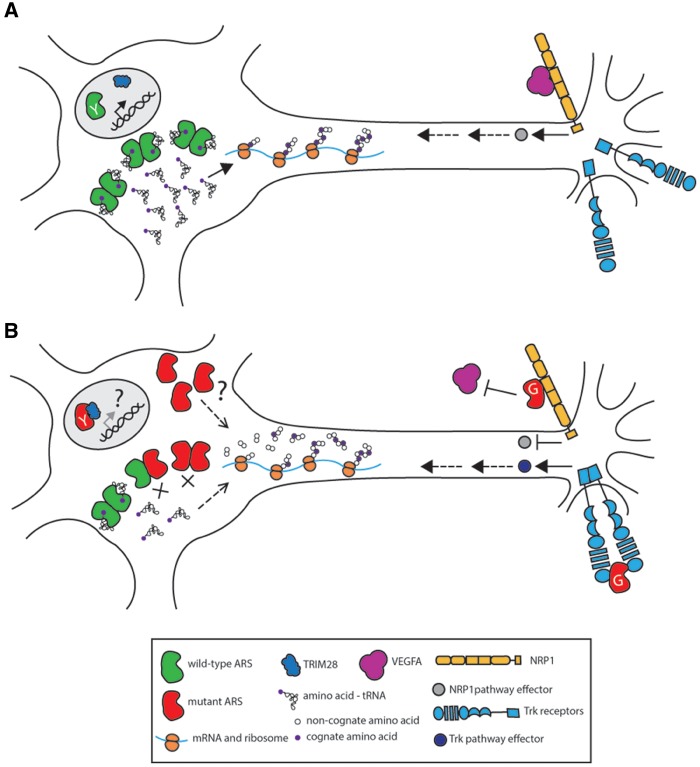
Aminoacyl-tRNA synthetases (ARSs) are responsible for charging amino acids to cognate tRNA molecules, which is the essential first step of protein translation. Interestingly, mutations in genes encoding ARS enzymes have been implicated in a broad spectrum of human inherited diseases. Bi-allelic mutations in ARSs typically cause severe, early-onset, recessive diseases that affect a wide range of tissues. The vast majority of these mutations show loss-of-function effects and impair protein translation. However, it is not clear how a subset cause tissue-specific phenotypes. In contrast, dominant ARS-mediated diseases specifically affect the peripheral nervous system-most commonly causing axonal peripheral neuropathy-and usually manifest later in life. These neuropathies are linked to heterozygosity for missense mutations in five ARS genes, which points to a shared mechanism of disease. However, it is not clear if a loss-of-function mechanism or a toxic gain-of-function mechanism is responsible for ARS-mediated neuropathy, or if a combination of these mechanisms operate on a mutation-specific basis. Here, we review our current understanding of recessive and dominant ARS-mediated disease. We also propose future directions for defining the molecular mechanisms of ARS mutations toward designing therapies for affected patient populations.
© The Author 2017. Published by Oxford University Press. All rights reserved. For Permissions, please email: journals.permissions@oup.com.
Figures
References
-
- Antonellis A., Green E.D. (2008) The role of aminoacyl-tRNA synthetases in genetic diseases. Annu. Rev. Genomics Hum. Genet., 9, 87–107. - PubMed
-
- Simons C., Griffin L.B., Helman G., Golas G., Pizzino A., Bloom M., Murphy J.L.P., Crawford J., Evans S.H., Topper S.. et al. (2015) Loss-of-function alanyl-tRNA synthetase mutations cause an autosomal-recessive early-onset epileptic encephalopathy with persistent myelination defect. Am. J. Hum. Genet., 96, 675–681. - PMC - PubMed
-
- Nakayama T., Wu J., Galvin-Parton P., Weiss J., Andriola M.R., Hill R.S., Vaughan D., El-Quessny M., Barry B.J., Partlow J.N.. et al. (2017) Deficient activity of alanyl-tRNA synthetase underlies an autosomal recessive syndrome of progressive microcephaly, hypomyelination, and epileptic encephalopathy. Hum. Mutat., doi: 10.1002/humu.23250, 10.1002/humu.23250. - PMC - PubMed
-
- Latour P., Thauvin-Robinet C., Baudelet-Méry C., Soichot P., Cusin V., Faivre L., Locatelli M.-C., Mayençon M., Sarcey A., Broussolle E.. et al. (2010) A major determinant for binding and aminoacylation of tRNAAla in cytoplasmic alanyl-tRNA synthetase is mutated in dominant axonal Charcot-Marie-tooth disease. Am. J. Hum. Genet., 86, 77–82. - PMC - PubMed
-
- McLaughlin H.M., Sakaguchi R., Giblin W., Wilson T.E., Biesecker L., Lupski J.R., Talbot K., Vance J.M., Züchner S., Lee Y.-C.. et al. (2012) A Recurrent loss‐of‐function alanyl‐tRNA synthetase (AARS ) mutation in patients with charcot‐marie‐tooth disease type 2N (CMT2N). Hum. Mutat., 33, 244–253. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
