

Inherited Paediatric Motor Neuron Disorders: Beyond Spinal Muscular Atrophy
- PMID: 28634552
- PMCID: PMC5467325
- DOI: 10.1155/2017/6509493
Inherited Paediatric Motor Neuron Disorders: Beyond Spinal Muscular Atrophy
Abstract
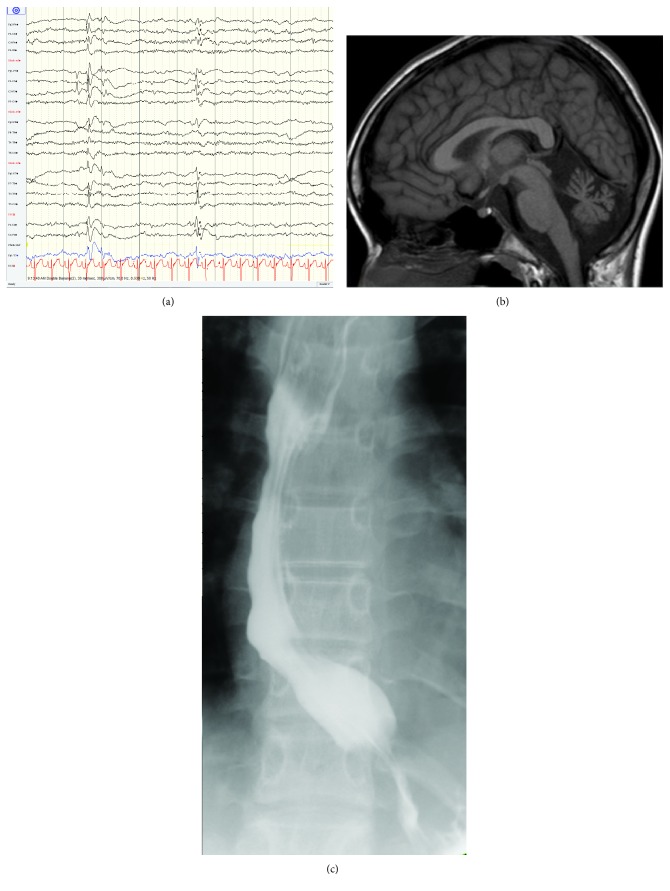
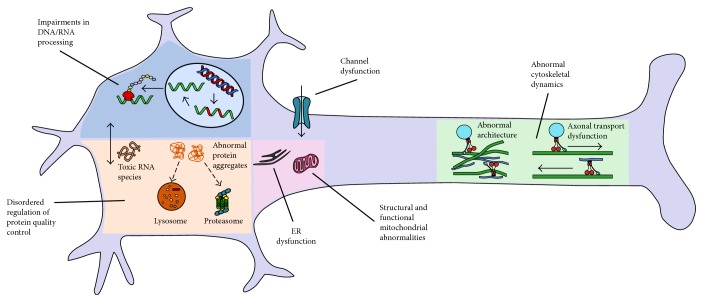
Paediatric motor neuron diseases encompass a group of neurodegenerative diseases characterised by the onset of muscle weakness and atrophy before the age of 18 years, attributable to motor neuron loss across various neuronal networks in the brain and spinal cord. While the genetic underpinnings are diverse, advances in next generation sequencing have transformed diagnostic paradigms. This has reinforced the clinical phenotyping and molecular genetic expertise required to navigate the complexities of such diagnoses. In turn, improved genetic technology and subsequent gene identification have enabled further insights into the mechanisms of motor neuron degeneration and how these diseases form part of a neurodegenerative disorder spectrum. Common pathophysiologies include abnormalities in axonal architecture and function, RNA processing, and protein quality control. This review incorporates an overview of the clinical manifestations, genetics, and pathophysiology of inherited paediatric motor neuron disorders beyond classic SMN1-related spinal muscular atrophy and describes recent advances in next generation sequencing and its clinical application. Specific disease-modifying treatment is becoming a clinical reality in some disorders of the motor neuron highlighting the importance of a timely and specific diagnosis.
Figures


References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
