

Structure-seq2: sensitive and accurate genome-wide profiling of RNA structure in vivo
- PMID: 28637286
- PMCID: PMC5737731
- DOI: 10.1093/nar/gkx533
Structure-seq2: sensitive and accurate genome-wide profiling of RNA structure in vivo
Abstract
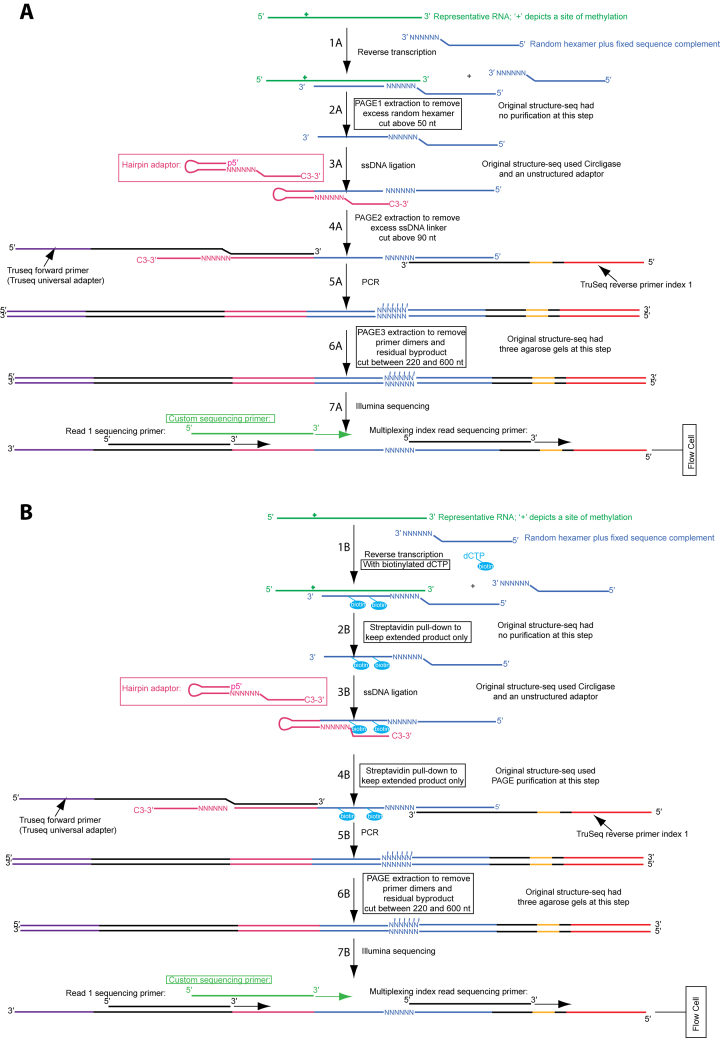
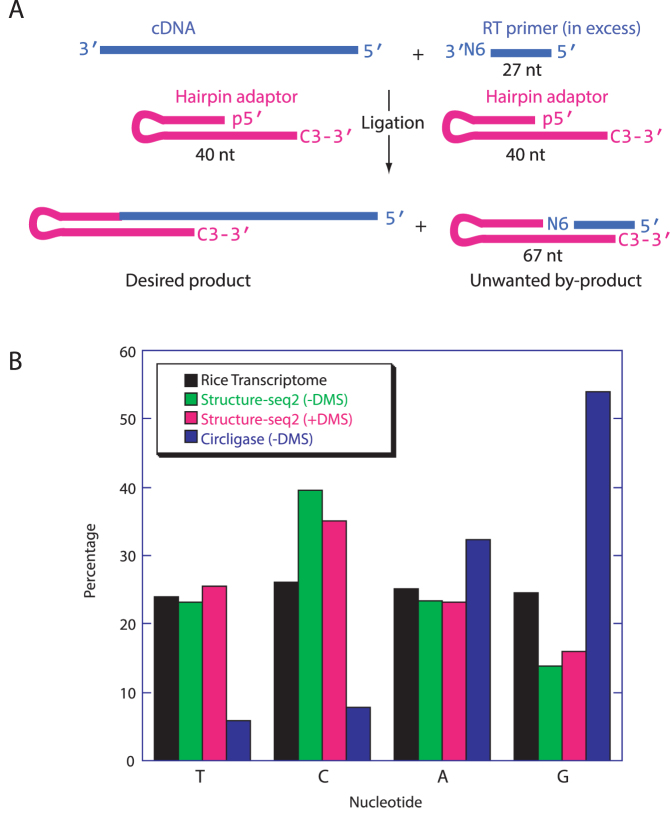
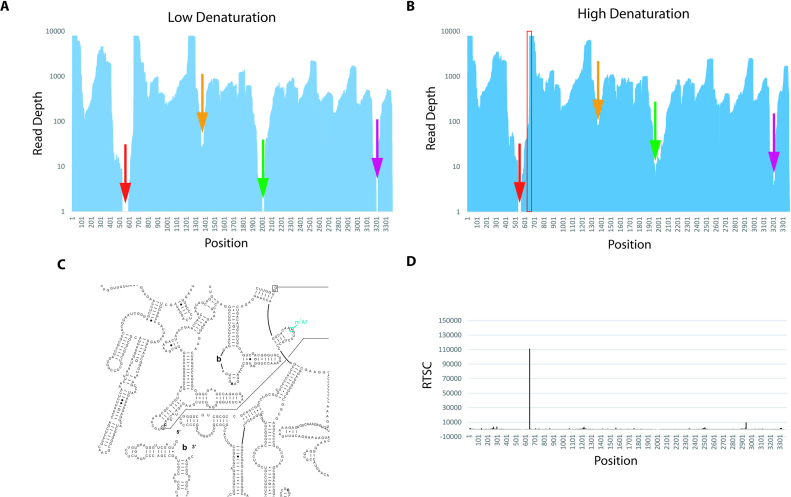
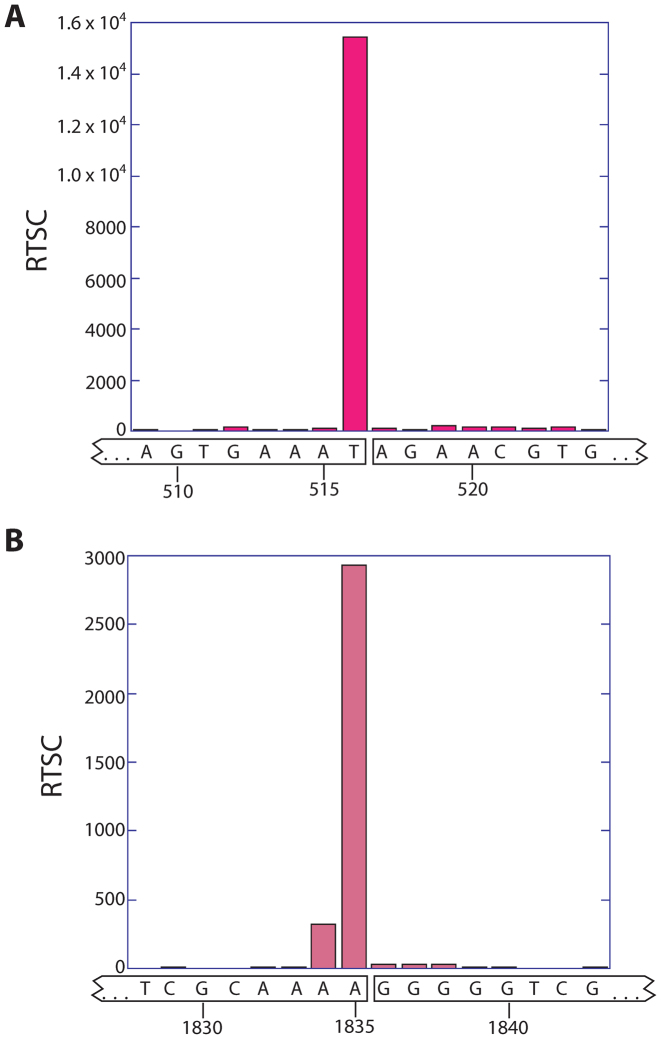
RNA serves many functions in biology such as splicing, temperature sensing, and innate immunity. These functions are often determined by the structure of RNA. There is thus a pressing need to understand RNA structure and how it changes during diverse biological processes both in vivo and genome-wide. Here, we present Structure-seq2, which provides nucleotide-resolution RNA structural information in vivo and genome-wide. This optimized version of our original Structure-seq method increases sensitivity by at least 4-fold and improves data quality by minimizing formation of a deleterious by-product, reducing ligation bias, and improving read coverage. We also present a variation of Structure-seq2 in which a biotinylated nucleotide is incorporated during reverse transcription, which greatly facilitates the protocol by eliminating two PAGE purification steps. We benchmark Structure-seq2 on both mRNA and rRNA structure in rice (Oryza sativa). We demonstrate that Structure-seq2 can lead to new biological insights. Our Structure-seq2 datasets uncover hidden breaks in chloroplast rRNA and identify a previously unreported N1-methyladenosine (m1A) in a nuclear-encoded Oryza sativa rRNA. Overall, Structure-seq2 is a rapid, sensitive, and unbiased method to probe RNA in vivo and genome-wide that facilitates new insights into RNA biology.
© The Author(s) 2017. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
References
-
- Bevilacqua P.C., Ritchey L.E., Su Z., Assmann S.M.. Genome-wide analysis of RNA secondary structure. Annu. Rev. Genet. 2016; 50:235–266. - PubMed
-
- Kwok C.K., Tang Y., Assmann S.M., Bevilacqua P.C.. The RNA structurome: transcriptome-wide structure probing with next-generation sequencing. Trends Biochem. Sci. 2015; 40:221–232. - PubMed
-
- Ding Y., Kwok C.K., Tang Y., Bevilacqua P.C., Assmann S.M.. Genome-wide profiling of in vivo RNA structure at single-nucleotide resolution using structure-seq. Nat. Protoc. 2015; 10:1050–1066. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
