

Same-day genomic and epigenomic diagnosis of brain tumors using real-time nanopore sequencing
- PMID: 28638988
- PMCID: PMC5645447
- DOI: 10.1007/s00401-017-1743-5
Same-day genomic and epigenomic diagnosis of brain tumors using real-time nanopore sequencing
Abstract
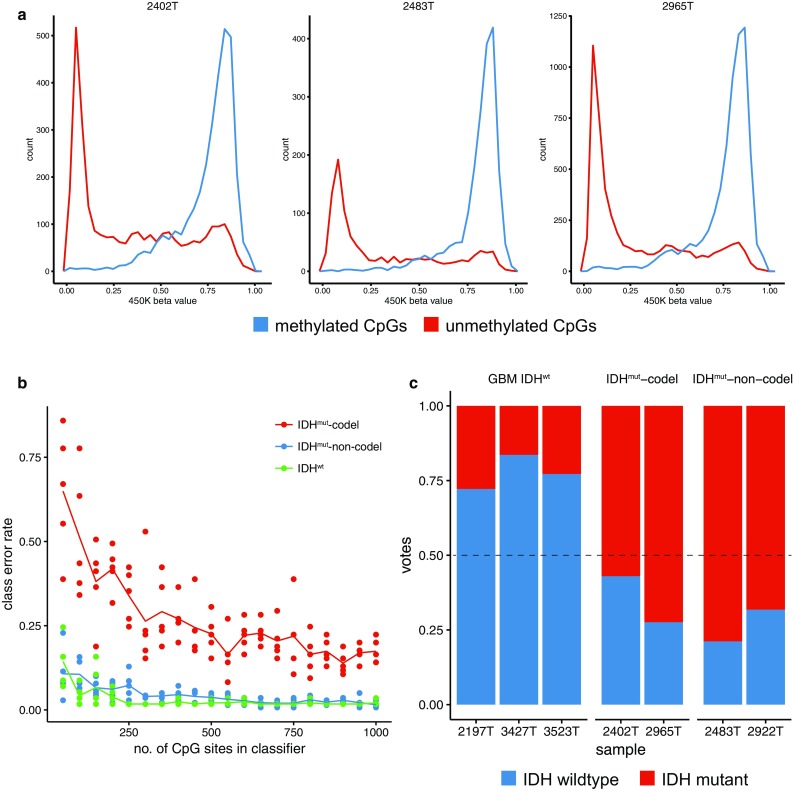
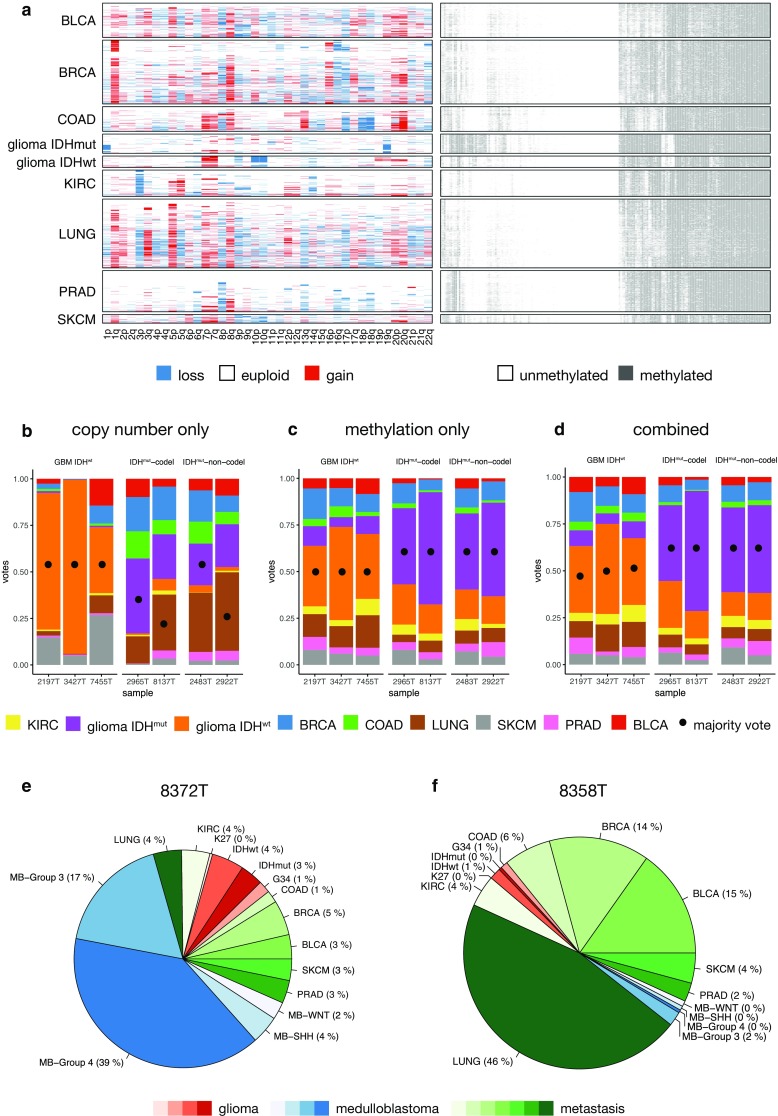
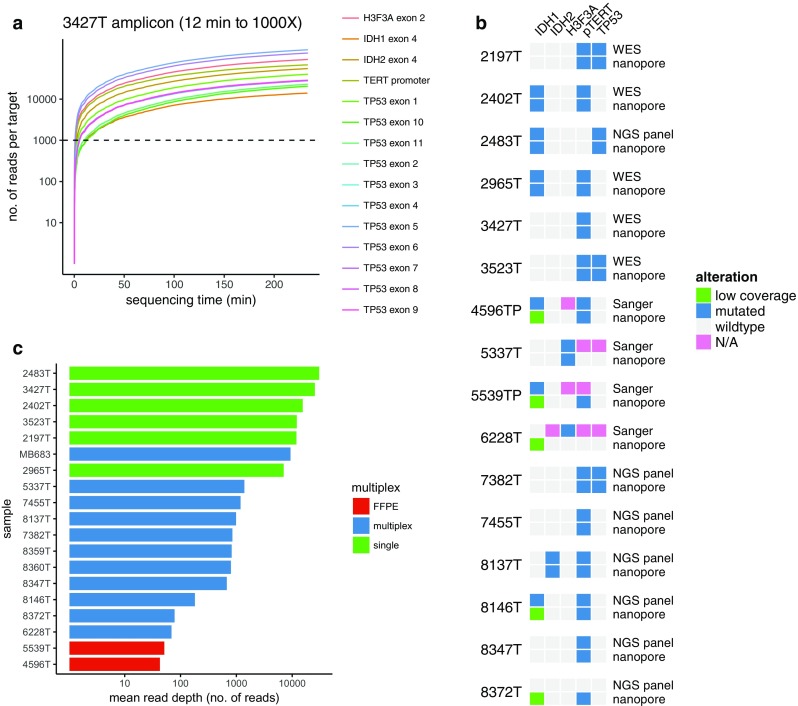
Molecular classification of cancer has entered clinical routine to inform diagnosis, prognosis, and treatment decisions. At the same time, new tumor entities have been identified that cannot be defined histologically. For central nervous system tumors, the current World Health Organization classification explicitly demands molecular testing, e.g., for 1p/19q-codeletion or IDH mutations, to make an integrated histomolecular diagnosis. However, a plethora of sophisticated technologies is currently needed to assess different genomic and epigenomic alterations and turnaround times are in the range of weeks, which makes standardized and widespread implementation difficult and hinders timely decision making. Here, we explored the potential of a pocket-size nanopore sequencing device for multimodal and rapid molecular diagnostics of cancer. Low-pass whole genome sequencing was used to simultaneously generate copy number (CN) and methylation profiles from native tumor DNA in the same sequencing run. Single nucleotide variants in IDH1, IDH2, TP53, H3F3A, and the TERT promoter region were identified using deep amplicon sequencing. Nanopore sequencing yielded ~0.1X genome coverage within 6 h and resulting CN and epigenetic profiles correlated well with matched microarray data. Diagnostically relevant alterations, such as 1p/19q codeletion, and focal amplifications could be recapitulated. Using ad hoc random forests, we could perform supervised pan-cancer classification to distinguish gliomas, medulloblastomas, and brain metastases of different primary sites. Single nucleotide variants in IDH1, IDH2, and H3F3A were identified using deep amplicon sequencing within minutes of sequencing. Detection of TP53 and TERT promoter mutations shows that sequencing of entire genes and GC-rich regions is feasible. Nanopore sequencing allows same-day detection of structural variants, point mutations, and methylation profiling using a single device with negligible capital cost. It outperforms hybridization-based and current sequencing technologies with respect to time to diagnosis and required laboratory equipment and expertise, aiming to make precision medicine possible for every cancer patient, even in resource-restricted settings.
Keywords: Brain tumor; Epigenomics; Glioma; Molecular neuropathology; Nanopore sequencing; Whole genome sequencing.
Conflict of interest statement
Funding
This work has been supported by Deutsche Forschungsgemeinschaft (EU 162/1-1 to PE), the program “Investissements d’avenir” (ANR-10-IAIHU-06 to AI), Institut Universitaire de Cancérologie (to AI), Ligue Nationale Contre le Cancer (to AI), Institut Carnot (to KL), and Fondation ARC pour la recherche sur le cancer (n°PJA 20151203562 to FB).
Conflict of interest
The authors declare no conflicts of interest.
Figures

References
-
- Breiman L. Random forests. Mach Learn. 2001;45:5–32. doi: 10.1023/A:1010933404324. - DOI
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
