

MicroRNA-34a dependent regulation of AXL controls the activation of dendritic cells in inflammatory arthritis
- PMID: 28639625
- PMCID: PMC5489689
- DOI: 10.1038/ncomms15877
MicroRNA-34a dependent regulation of AXL controls the activation of dendritic cells in inflammatory arthritis
Abstract
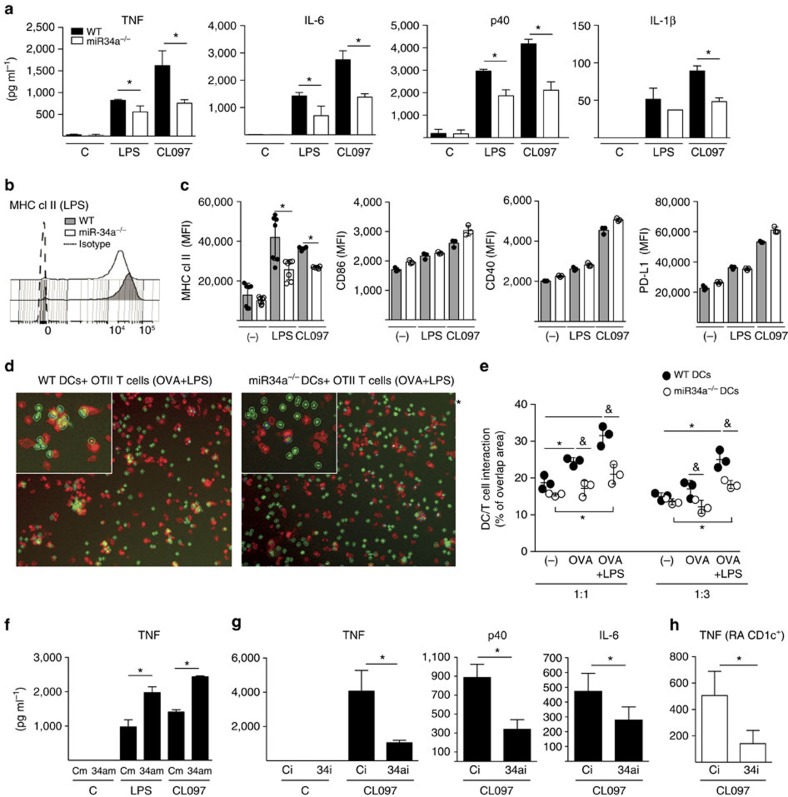
Current treatments for rheumatoid arthritis (RA) do not reverse underlying aberrant immune function. A genetic predisposition to RA, such as HLA-DR4 positivity, indicates that dendritic cells (DC) are of crucial importance to pathogenesis by activating auto-reactive lymphocytes. Here we show that microRNA-34a provides homoeostatic control of CD1c+ DC activation via regulation of tyrosine kinase receptor AXL, an important inhibitory DC auto-regulator. This pathway is aberrant in CD1c+ DCs from patients with RA, with upregulation of miR-34a and lower levels of AXL compared to DC from healthy donors. Production of pro-inflammatory cytokines is reduced by ex vivo gene-silencing of miR-34a. miR-34a-deficient mice are resistant to collagen-induced arthritis and interaction of DCs and T cells from these mice are reduced and do not support the development of Th17 cells in vivo. Our findings therefore show that miR-34a is an epigenetic regulator of DC function that may contribute to RA.
Conflict of interest statement
The authors declare no competing financial interests.
Figures






References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
