

Drug-Target Kinetics in Drug Discovery
- PMID: 28640596
- PMCID: PMC5767540
- DOI: 10.1021/acschemneuro.7b00185
Drug-Target Kinetics in Drug Discovery
Abstract
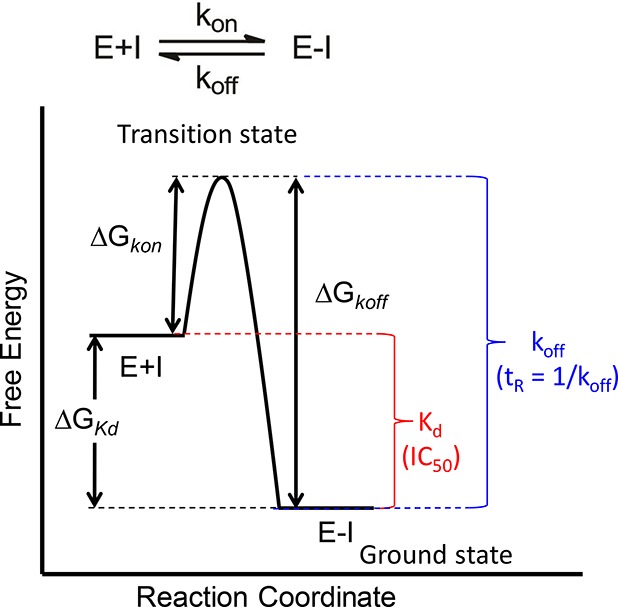
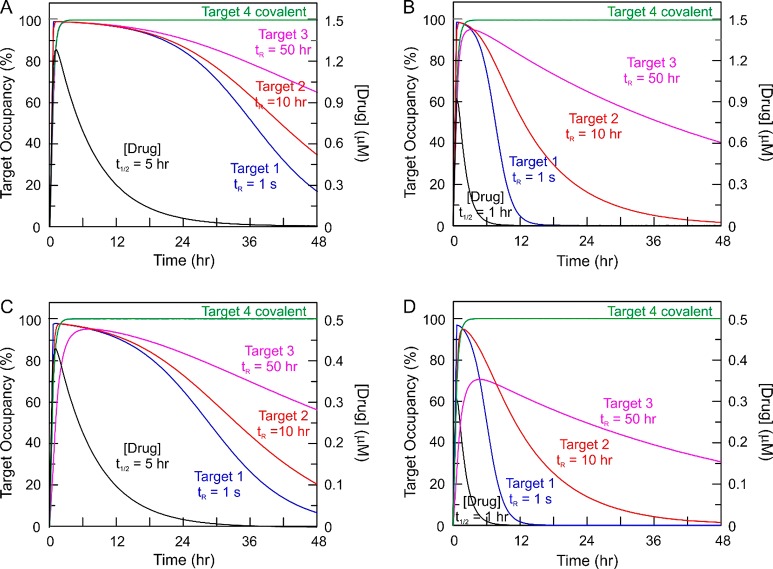
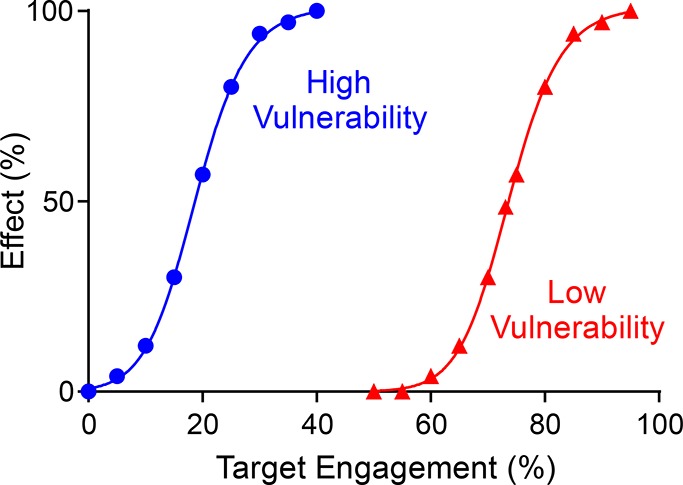
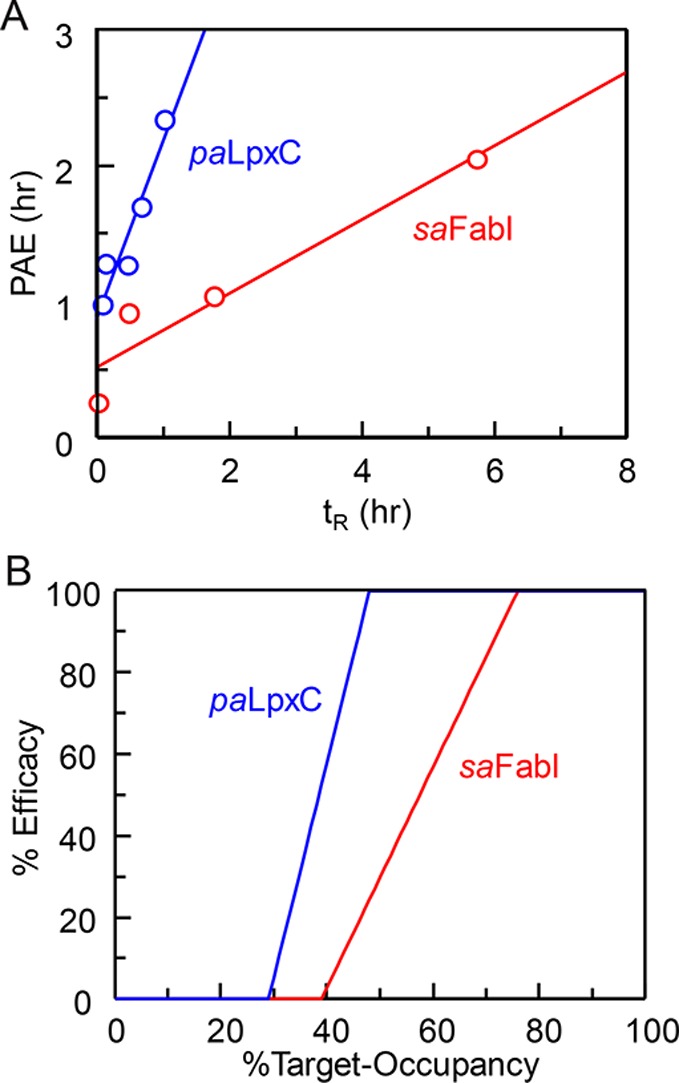
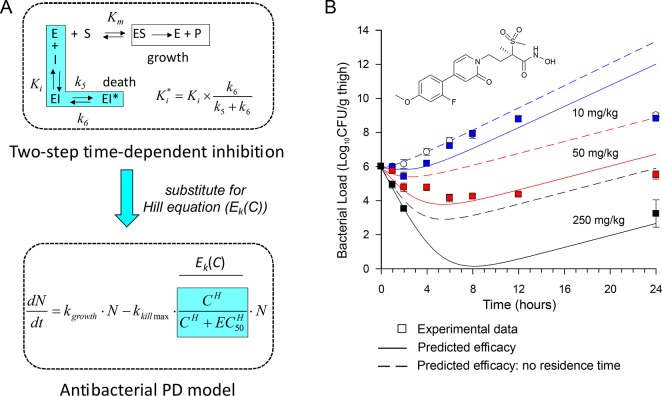
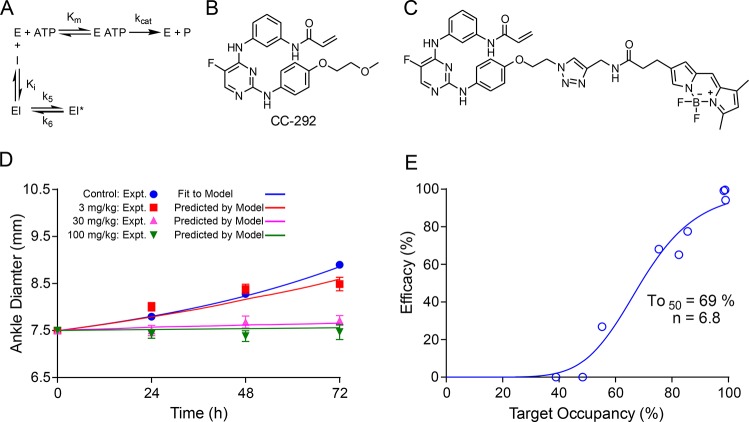
The development of therapies for the treatment of neurological cancer faces a number of major challenges including the synthesis of small molecule agents that can penetrate the blood-brain barrier (BBB). Given the likelihood that in many cases drug exposure will be lower in the CNS than in systemic circulation, it follows that strategies should be employed that can sustain target engagement at low drug concentration. Time dependent target occupancy is a function of both the drug and target concentration as well as the thermodynamic and kinetic parameters that describe the binding reaction coordinate, and sustained target occupancy can be achieved through structural modifications that increase target (re)binding and/or that decrease the rate of drug dissociation. The discovery and deployment of compounds with optimized kinetic effects requires information on the structure-kinetic relationships that modulate the kinetics of binding, and the molecular factors that control the translation of drug-target kinetics to time-dependent drug activity in the disease state. This Review first introduces the potential benefits of drug-target kinetics, such as the ability to delineate both thermodynamic and kinetic selectivity, and then describes factors, such as target vulnerability, that impact the utility of kinetic selectivity. The Review concludes with a description of a mechanistic PK/PD model that integrates drug-target kinetics into predictions of drug activity.
Keywords: CNS cancer; Kinetic selectivity; PK/PD modeling; blood brain barrier; drug-target residence time; target occupancy; target vulnerability; therapeutic window.
Conflict of interest statement
The author declares no competing financial interest.
Figures
References
-
- Levin V. A.; Tonge P. J.; Gallo J. M.; Birtwistle M. R.; Dar A. C.; Iavarone A.; Paddison P. J.; Heffron T. P.; Elmquist W. F.; Lachowicz J. E.; Johnson T. W.; White F. M.; Sul J.; Smith Q. R.; Shen W.; Sarkaria J. N.; Samala R.; Wen P. Y.; Berry D. A.; Petter R. C. (2015) CNS Anticancer Drug Discovery and Development Conference White Paper. Neuro-Oncology 17 (Suppl 6), vi1–vi26. 10.1093/neuonc/nov169. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
