

Targeting Microglial Activation States as a Therapeutic Avenue in Parkinson's Disease
- PMID: 28642697
- PMCID: PMC5463358
- DOI: 10.3389/fnagi.2017.00176
Targeting Microglial Activation States as a Therapeutic Avenue in Parkinson's Disease
Abstract
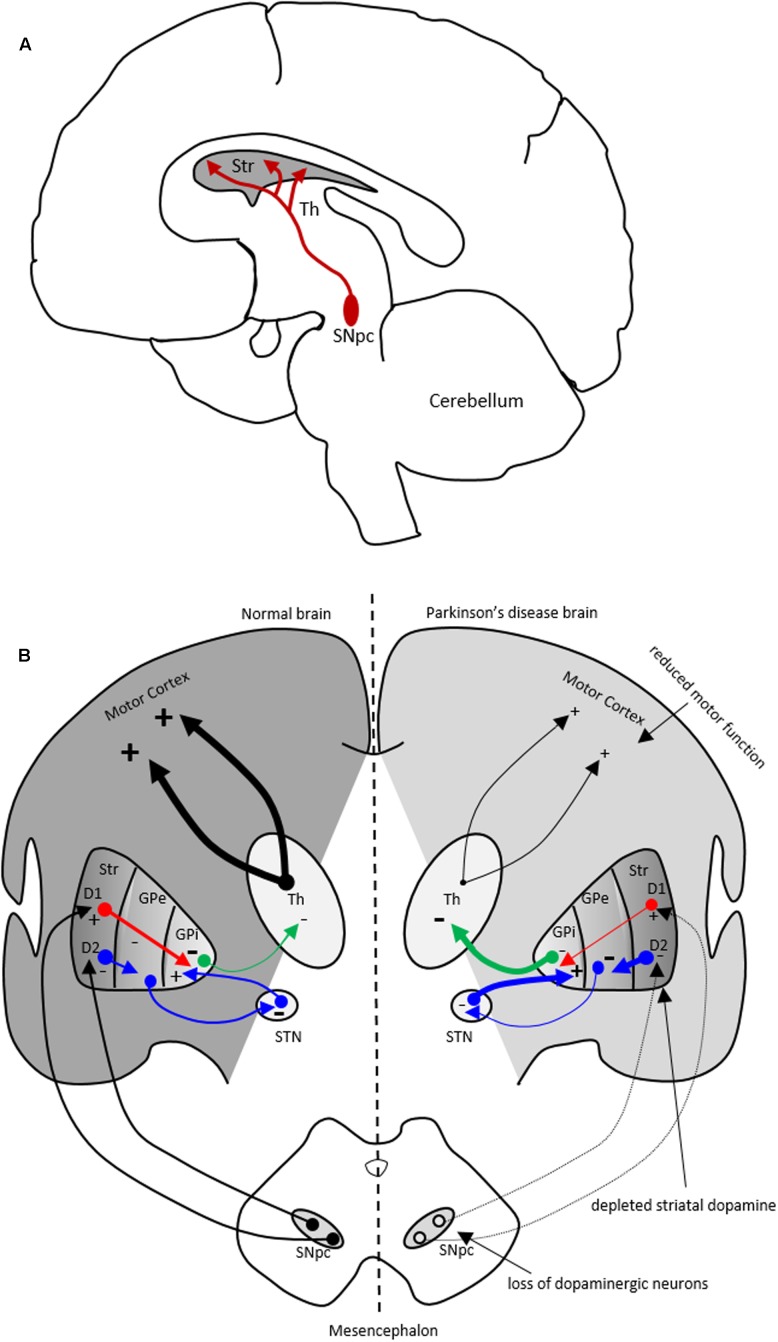
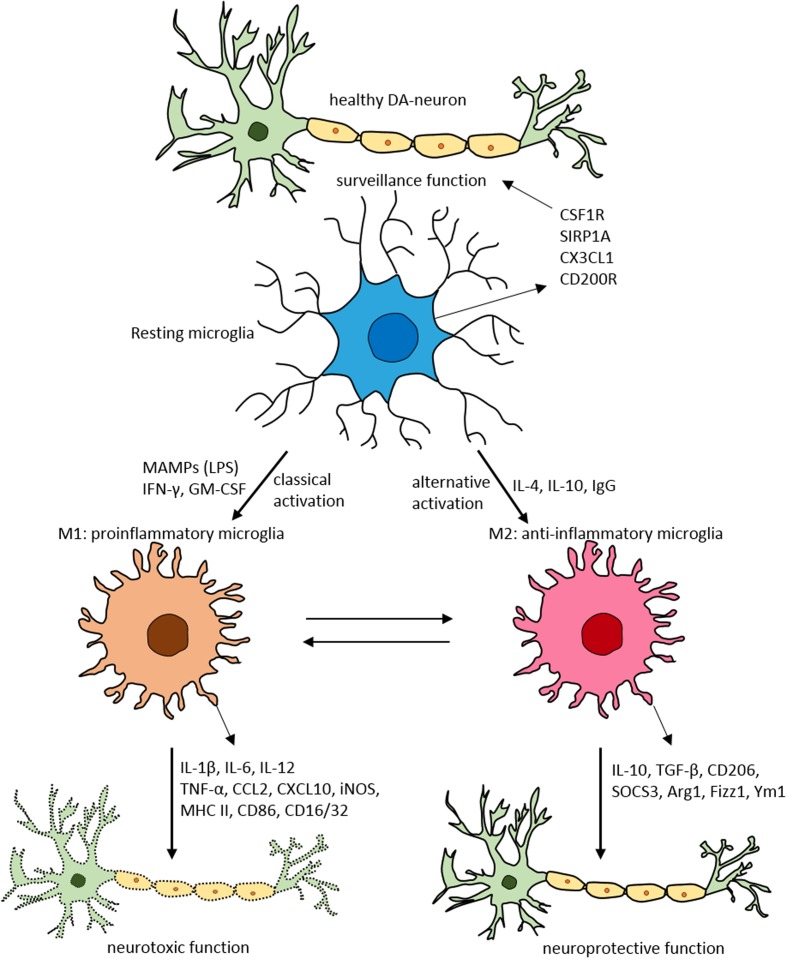
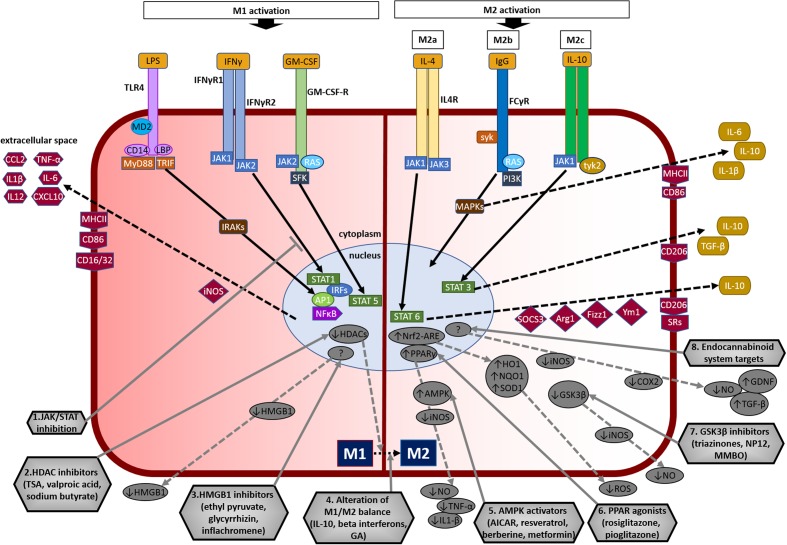
Parkinson's disease (PD) is a chronic and progressive disorder characterized neuropathologically by loss of dopamine neurons in the substantia nigra, intracellular proteinaceous inclusions, reduction of dopaminergic terminals in the striatum, and increased neuroinflammatory cells. The consequent reduction of dopamine in the basal ganglia results in the classical parkinsonian motor phenotype. A growing body of evidence suggest that neuroinflammation mediated by microglia, the resident macrophage-like immune cells in the brain, play a contributory role in PD pathogenesis. Microglia participate in both physiological and pathological conditions. In the former, microglia restore the integrity of the central nervous system and, in the latter, they promote disease progression. Microglia acquire different activation states to modulate these cellular functions. Upon activation to the M1 phenotype, microglia elaborate pro-inflammatory cytokines and neurotoxic molecules promoting inflammation and cytotoxic responses. In contrast, when adopting the M2 phenotype microglia secrete anti-inflammatory gene products and trophic factors that promote repair, regeneration, and restore homeostasis. Relatively little is known about the different microglial activation states in PD and a better understanding is essential for developing putative neuroprotective agents. Targeting microglial activation states by suppressing their deleterious pro-inflammatory neurotoxicity and/or simultaneously enhancing their beneficial anti-inflammatory protective functions appear as a valid therapeutic approach for PD treatment. In this review, we summarize microglial functions and, their dual neurotoxic and neuroprotective role in PD. We also review molecules that modulate microglial activation states as a therapeutic option for PD treatment.
Keywords: Parkinson’s disease; microglia; neuroinflammation; polarization; therapeutics.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources