

An Unexplained Congenital Disorder of Glycosylation-II in a Child with Neurohepatic Involvement, Hypercholesterolemia and Hypoceruloplasminemia
- PMID: 28643274
- PMCID: PMC5874206
- DOI: 10.1007/8904_2017_35
An Unexplained Congenital Disorder of Glycosylation-II in a Child with Neurohepatic Involvement, Hypercholesterolemia and Hypoceruloplasminemia
Abstract
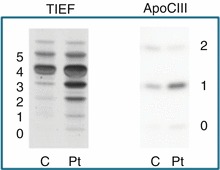
We report on a 12-year-old adopted boy with psychomotor disability, absence seizures, and normal brain MRI. He showed increased (but initially, at 5 months, normal) serum cholesterol, increased alkaline phosphatases, transiently increased transaminases and hypoceruloplasminemia with normal serum and urinary copper. Blood levels of immunoglobulins, haptoglobin, antithrombin, and factor XI were normal. A type 2 serum transferrin isoelectrofocusing and hypoglycosylation of apoCIII pointed to a combined N- and O-glycosylation defect. Neither CDG panel analysis with 79 CDG-related genes, nor whole exome sequencing revealed the cause of this CDG. Whole genome sequencing was not performed since the biological parents of this adopted child were not available.
Keywords: CDG-II; Hypercholesterolemia; Hypoceruloplasminemia; Neurohepatic involvement.
Figures
References
-
- Harrison H, Miller K. Multiple serum protein abnormalities in carbohydrate-deficient glycoprotein syndrome: pathognomonic finding of two-dimensional electrophoresis. Clin Chem. 1992;38:1390–1392. - PubMed
-
- Henri H, Tissot JD, Messerli B, et al. Microheterogeneity of serum glycoproteins and their liver precursors in patients with carbohydrate-deficient glycoprotein syndrome type I: apparent deficiencies in clusterin and serum amyloid P. J Lab Clin Med. 1997;129:412–421. doi: 10.1016/S0022-2143(97)90074-3. - DOI - PubMed
-
- Jaeken J, Morava E (2016) Congenital disorders of glycosylation, dolichol and glycosylphosphatidylinositol metabolism. In: Saudubray JM, van den Berghe G, Walter JH (eds) Inborn metabolic diseases – diagnosis and treatment, 6th edn. Springer, Berlin
LinkOut - more resources
Full Text Sources
Other Literature Sources
