

New protein structures provide an updated understanding of phenylketonuria
- PMID: 28645531
- PMCID: PMC5549558
- DOI: 10.1016/j.ymgme.2017.06.005
New protein structures provide an updated understanding of phenylketonuria
Abstract
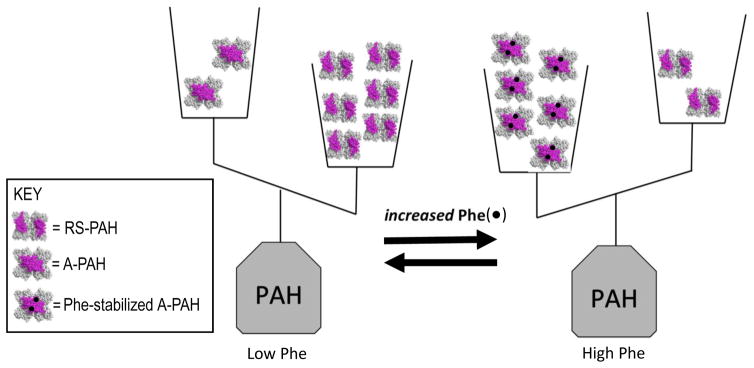
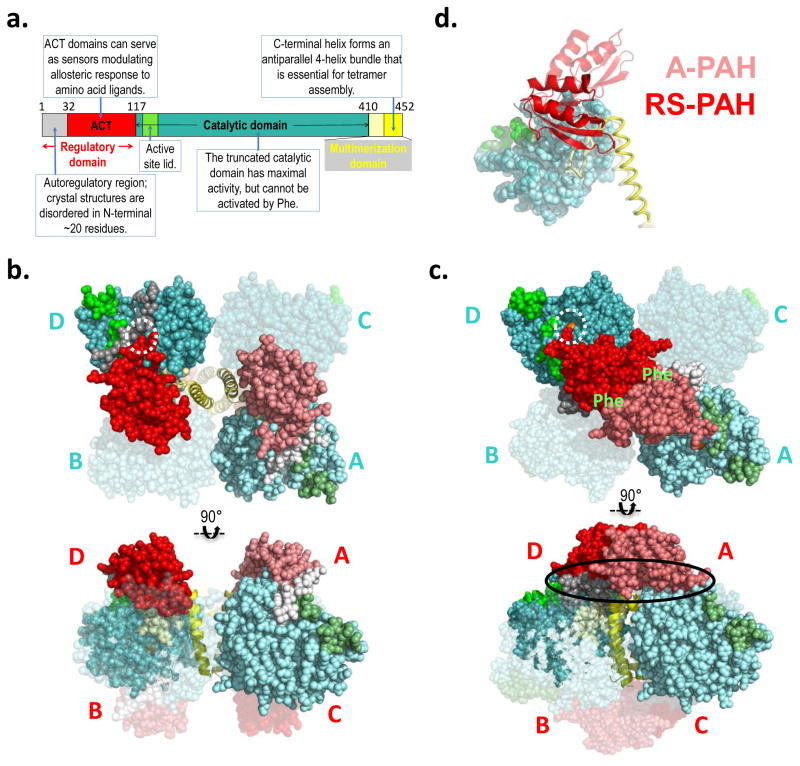
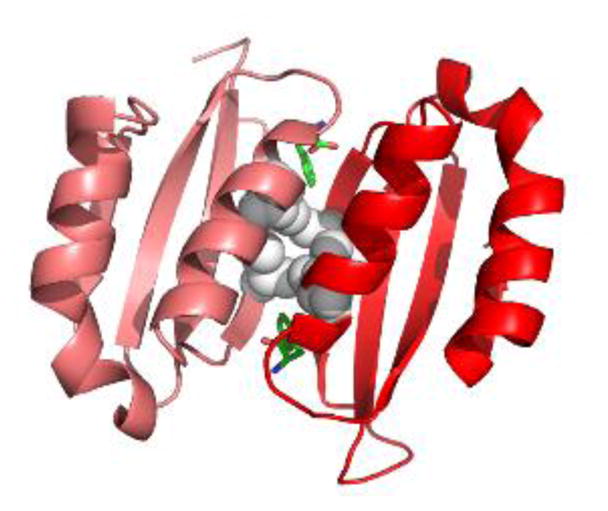
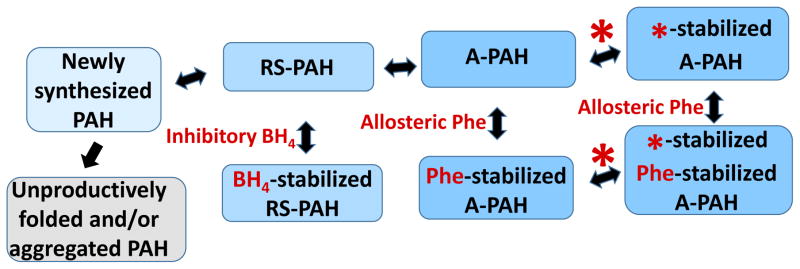
Phenylketonuria (PKU) and less severe hyperphenylalaninemia (HPA) constitute the most common inborn error of amino acid metabolism, and is most often caused by defects in phenylalanine hydroxylase (PAH) function resulting in accumulation of Phe to neurotoxic levels. Despite the success of dietary intervention in preventing permanent neurological damage, individuals living with PKU clamor for additional non-dietary therapies. The bulk of disease-associated mutations are PAH missense variants, which occur throughout the entire 452 amino acid human PAH protein. While some disease-associated mutations affect protein structure (e.g. truncations) and others encode catalytically dead variants, most have been viewed as defective in protein folding/stability. Here we refine this view to address how PKU-associated missense variants can perturb the equilibrium among alternate native PAH structures (resting-state PAH and activated PAH), thus shifting the tipping point of this equilibrium to a neurotoxic Phe concentration. This refined view of PKU introduces opportunities for the design or discovery of therapeutic pharmacological chaperones that can help restore the tipping point to healthy Phe levels and how such a therapeutic might work with or without the inhibitory pharmacological chaperone BH4. Dysregulation of an equilibrium of architecturally distinct native PAH structures departs from the concept of "misfolding", provides an updated understanding of PKU, and presents an enhanced foundation for understanding genotype/phenotype relationships.
Keywords: Allostery; Conformational selection; Pharmacological chaperones; Phenylalanine hydroxylase; Phenylketonuria.
Copyright © 2017 Elsevier Inc. All rights reserved.
Figures
References
-
- Jahja R, Huijbregts SC, de Sonneville LM, van der Meere JJ, van Spronsen FJ. Neurocognitive evidence for revision of treatment targets and guidelines for phenylketonuria. J Pediatr. 2014;164(4):895–899. e892. - PubMed
-
- Greene CL, Longo N. National Institutes of Health (NIH) review of evidence in phenylalanine hydroxylase deficiency (phenylketonuria) and recommendations/guidelines for therapy from the American College of Medical Genetics (ACMG) and Genetics Metabolic Dietitians International (GMDI) Mol Genet Metab. 2014;112(2):85–86. - PubMed
-
- Camp KM, et al. Phenylketonuria Scientific Review Conference: state of the science and future research needs. Mol Genet Metab. 2014;112(2):87–122. - PubMed
-
- van Spronsen FJ. Phenylketonuria management from an European perspective: a commentary. Mol Genet Metab. 2010;100(2):107–110. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
