

CRISPR-Cas12a-Assisted Recombineering in Bacteria
- PMID: 28646112
- PMCID: PMC5561284
- DOI: 10.1128/AEM.00947-17
CRISPR-Cas12a-Assisted Recombineering in Bacteria
Abstract
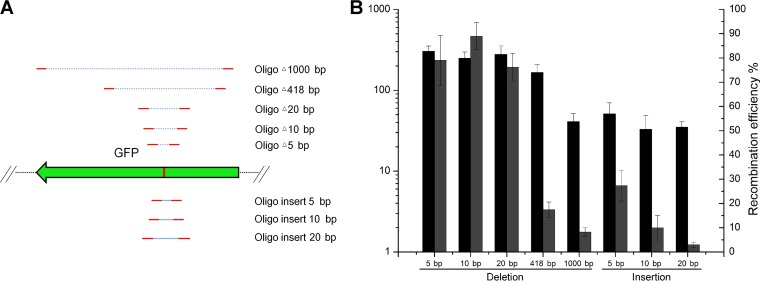
Clustered regularly interspaced short palindromic repeat (CRISPR)-Cas12a (Cpf1) has emerged as an effective genome editing tool in many organisms. Here, we developed and optimized a CRISPR-Cas12a-assisted recombineering system to facilitate genetic manipulation in bacteria. Using this system, point mutations, deletions, insertions, and gene replacements can be easily generated on the chromosome or native plasmids in Escherichia coli, Yersinia pestis, and Mycobacterium smegmatis Because CRISPR-Cas12a-assisted recombineering does not require introduction of an antibiotic resistance gene into the chromosome to select for recombinants, it is an efficient approach for generating markerless and scarless mutations in bacteria.IMPORTANCE The CRISPR-Cas9 system has been widely used to facilitate genome editing in many bacteria. CRISPR-Cas12a (Cpf1), a new type of CRISPR-Cas system, allows efficient genome editing in bacteria when combined with recombineering. Cas12a and Cas9 recognize different target sites, which allows for more precise selection of the cleavage target and introduction of the desired mutation. In addition, CRISPR-Cas12a-assisted recombineering can be used for genetic manipulation of plasmids and plasmid curing. Finally, Cas12a-assisted recombineering in the generation of point mutations, deletions, insertions, and replacements in bacteria has been systematically analyzed. Taken together, our findings will guide efficient Cas12a-mediated genome editing in bacteria.
Keywords: Cas12a; Mycobacterium smegmatis; Yersinia pestis; recombineering.
Copyright © 2017 Yan et al.
Figures





References
-
- Makarova KS, Wolf YI, Alkhnbashi OS, Costa F, Shah SA, Saunders SJ, Barrangou R, Brouns SJ, Charpentier E, Haft DH, Horvath P, Moineau S, Mojica FJ, Terns RM, Terns MP, White MF, Yakunin AF, Garrett RA, van der Oost J, Backofen R, Koonin EV. 2015. An updated evolutionary classification of CRISPR-Cas systems. Nat Rev Microbiol 13:722–736. doi: 10.1038/nrmicro3569. - DOI - PMC - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
