

CMSA: a heterogeneous CPU/GPU computing system for multiple similar RNA/DNA sequence alignment
- PMID: 28646874
- PMCID: PMC5483318
- DOI: 10.1186/s12859-017-1725-6
CMSA: a heterogeneous CPU/GPU computing system for multiple similar RNA/DNA sequence alignment
Abstract
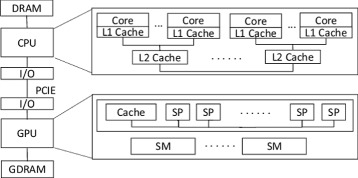
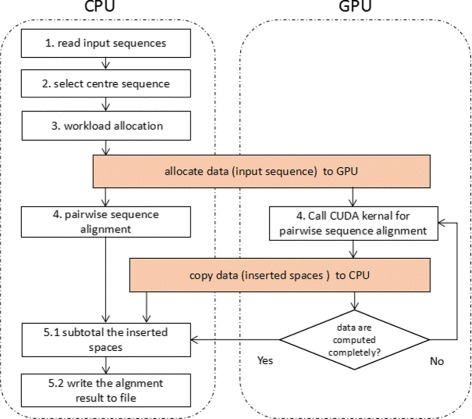
Background: The multiple sequence alignment (MSA) is a classic and powerful technique for sequence analysis in bioinformatics. With the rapid growth of biological datasets, MSA parallelization becomes necessary to keep its running time in an acceptable level. Although there are a lot of work on MSA problems, their approaches are either insufficient or contain some implicit assumptions that limit the generality of usage. First, the information of users' sequences, including the sizes of datasets and the lengths of sequences, can be of arbitrary values and are generally unknown before submitted, which are unfortunately ignored by previous work. Second, the center star strategy is suited for aligning similar sequences. But its first stage, center sequence selection, is highly time-consuming and requires further optimization. Moreover, given the heterogeneous CPU/GPU platform, prior studies consider the MSA parallelization on GPU devices only, making the CPUs idle during the computation. Co-run computation, however, can maximize the utilization of the computing resources by enabling the workload computation on both CPU and GPU simultaneously.
Results: This paper presents CMSA, a robust and efficient MSA system for large-scale datasets on the heterogeneous CPU/GPU platform. It performs and optimizes multiple sequence alignment automatically for users' submitted sequences without any assumptions. CMSA adopts the co-run computation model so that both CPU and GPU devices are fully utilized. Moreover, CMSA proposes an improved center star strategy that reduces the time complexity of its center sequence selection process from O(mn 2) to O(mn). The experimental results show that CMSA achieves an up to 11× speedup and outperforms the state-of-the-art software.
Conclusion: CMSA focuses on the multiple similar RNA/DNA sequence alignment and proposes a novel bitmap based algorithm to improve the center star strategy. We can conclude that harvesting the high performance of modern GPU is a promising approach to accelerate multiple sequence alignment. Besides, adopting the co-run computation model can maximize the entire system utilization significantly. The source code is available at https://github.com/wangvsa/CMSA .
Keywords: Center star alignment; GPU; Heterogeneous; Multiple sequence alignment (MSA).
Figures

References
-
- Karadimitriou K, Kraft DH. Genetic algorithms and the multiple sequence alignment problem in biology. In: Proceedings of the Second Annual Molecular Biology and Biotechnology Conference. Baton Rouge: 1996. p. 1–7.
-
- Zou Q, Shan X, Jiang Y. A novel center star multiple sequence alignment algorithm based on affine gap penalty and k-band. Phys Procedia. 2012;33:322–7. doi: 10.1016/j.phpro.2012.05.069. - DOI
-
- Wang J, Guo M, Liu X, Liu Y, Wang C, Xing L, Che K. Lnetwork: an efficient and effective method for constructing phylogenetic networks. Bioinformatics. 2013;29:378. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
