

NAD(H) and NADP(H) Redox Couples and Cellular Energy Metabolism
- PMID: 28648096
- PMCID: PMC5737637
- DOI: 10.1089/ars.2017.7216
NAD(H) and NADP(H) Redox Couples and Cellular Energy Metabolism
Abstract
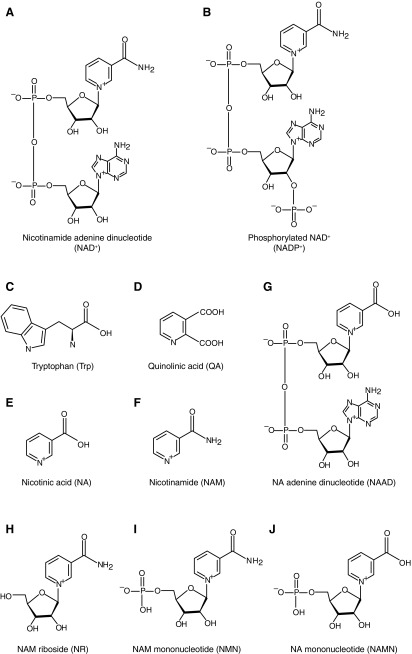
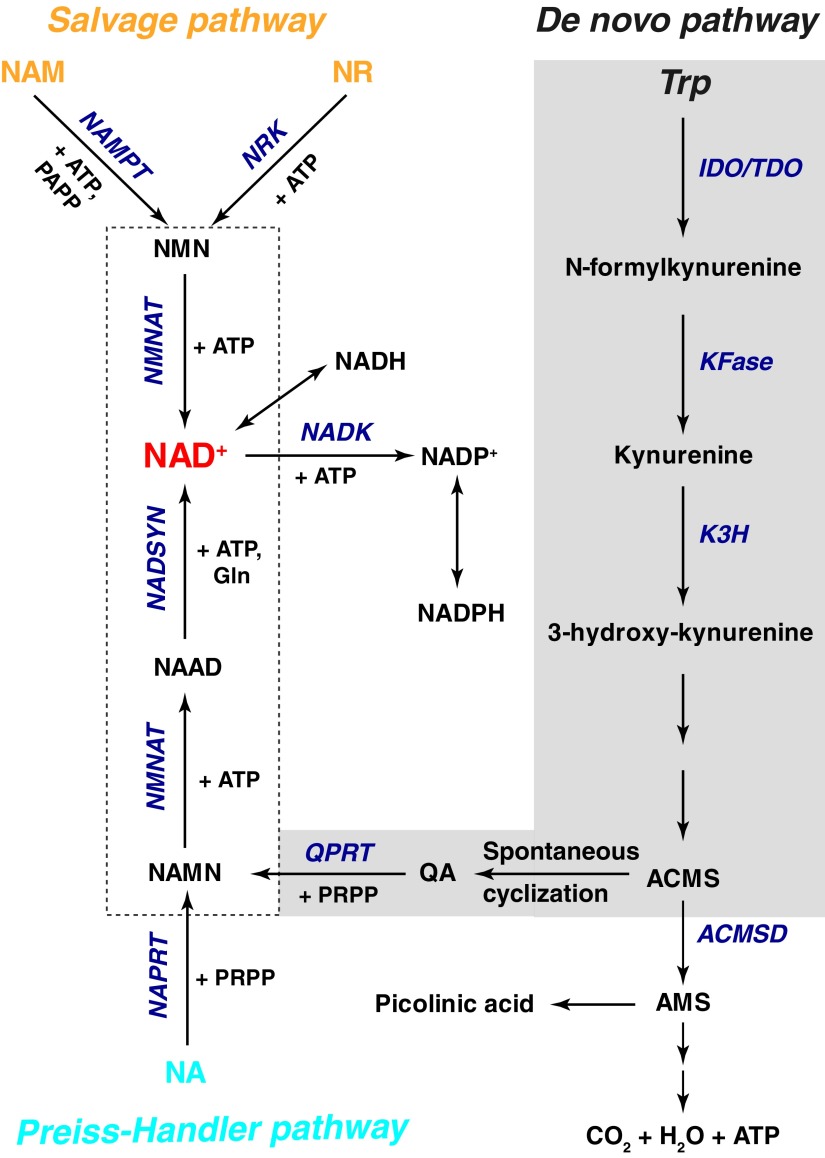
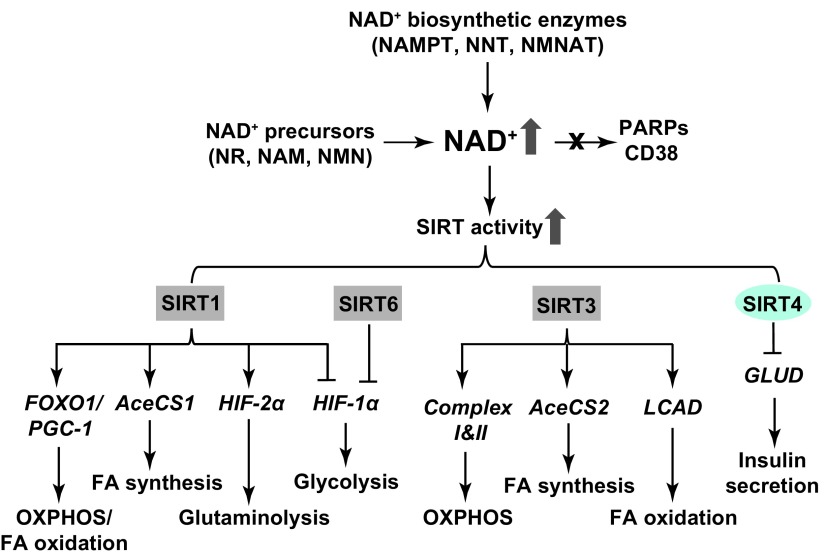
Significance: The nicotinamide adenine dinucleotide (NAD+)/reduced NAD+ (NADH) and NADP+/reduced NADP+ (NADPH) redox couples are essential for maintaining cellular redox homeostasis and for modulating numerous biological events, including cellular metabolism. Deficiency or imbalance of these two redox couples has been associated with many pathological disorders. Recent Advances: Newly identified biosynthetic enzymes and newly developed genetically encoded biosensors enable us to understand better how cells maintain compartmentalized NAD(H) and NADP(H) pools. The concept of redox stress (oxidative and reductive stress) reflected by changes in NAD(H)/NADP(H) has increasingly gained attention. The emerging roles of NAD+-consuming proteins in regulating cellular redox and metabolic homeostasis are active research topics.
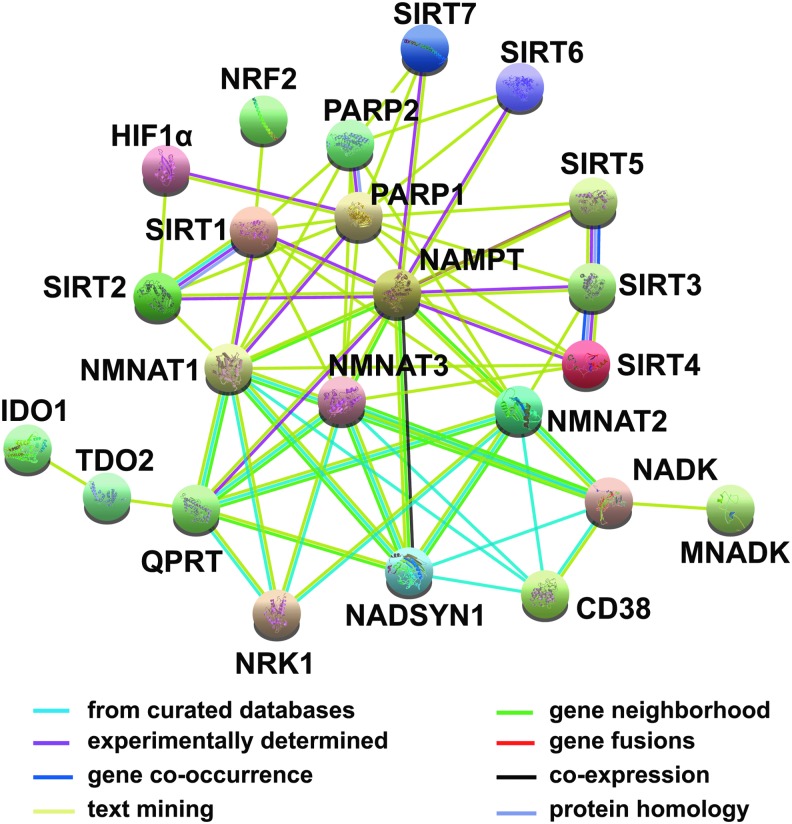
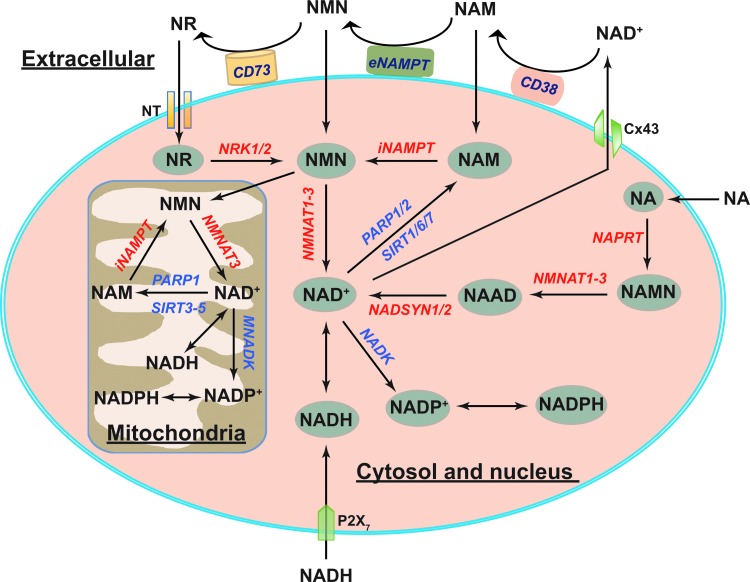
Critical issues: The biosynthesis and distribution of cellular NAD(H) and NADP(H) are highly compartmentalized. It is critical to understand how cells maintain the steady levels of these redox couple pools to ensure their normal functions and simultaneously avoid inducing redox stress. In addition, it is essential to understand how NAD(H)- and NADP(H)-utilizing enzymes interact with other signaling pathways, such as those regulated by hypoxia-inducible factor, to maintain cellular redox homeostasis and energy metabolism.
Future directions: Additional studies are needed to investigate the inter-relationships among compartmentalized NAD(H)/NADP(H) pools and how these two dinucleotide redox couples collaboratively regulate cellular redox states and cellular metabolism under normal and pathological conditions. Furthermore, recent studies suggest the utility of using pharmacological interventions or nutrient-based bioactive NAD+ precursors as therapeutic interventions for metabolic diseases. Thus, a better understanding of the cellular functions of NAD(H) and NADP(H) may facilitate efforts to address a host of pathological disorders effectively. Antioxid. Redox Signal. 28, 251-272.
Keywords: NAD(H); NADP(H); cellular metabolism; oxidative stress; redox state; reductive stress.
Figures
References
-
- Adam-Vizi V. and Chinopoulos C. Bioenergetics and the formation of mitochondrial reactive oxygen species. Trends Pharmacol Sci 27: 639–645, 2006 - PubMed
-
- Aksoy P, Escande C, White TA, Thompson M, Soares S, Benech JC, and Chini EN. Regulation of SIRT 1 mediated NAD dependent deacetylation: A novel role for the multifunctional enzyme CD38. Biochem Biophys Res Commun 349: 353–359, 2006 - PubMed
-
- Alberati-Giani D, Cesura AM, Broger C, Warren WD, Rover S, and Malherbe P. Cloning and functional expression of human kynurenine 3-monooxygenase. FEBS Lett 410: 407–412, 1997 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
