

Clostridium chauvoei, an Evolutionary Dead-End Pathogen
- PMID: 28649238
- PMCID: PMC5465433
- DOI: 10.3389/fmicb.2017.01054
Clostridium chauvoei, an Evolutionary Dead-End Pathogen
Erratum in
-
Corrigendum: Clostridium chauvoei, an Evolutionary Dead-End Pathogen.Front Microbiol. 2018 Mar 6;9:421. doi: 10.3389/fmicb.2018.00421. eCollection 2018. Front Microbiol. 2018. PMID: 29531522 Free PMC article.
Abstract
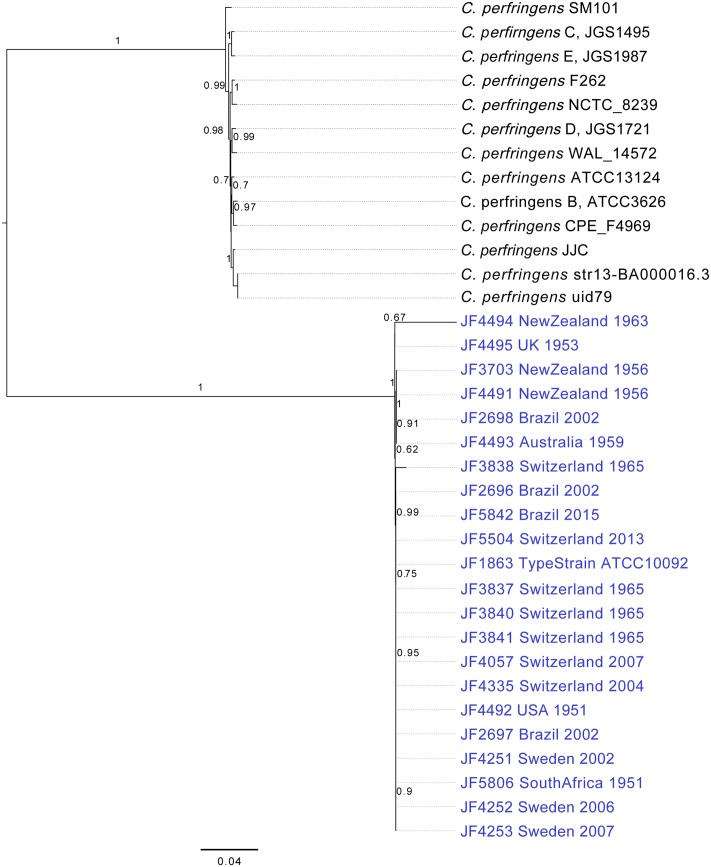
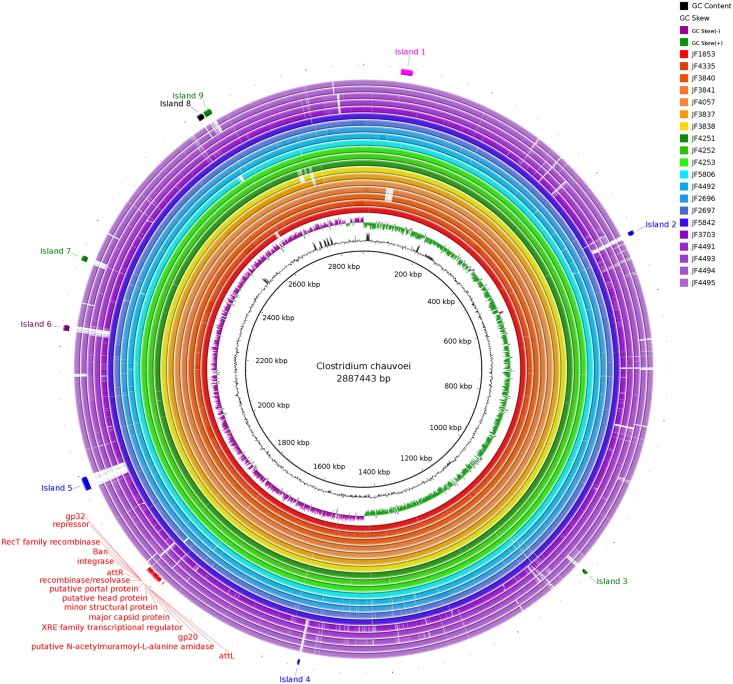
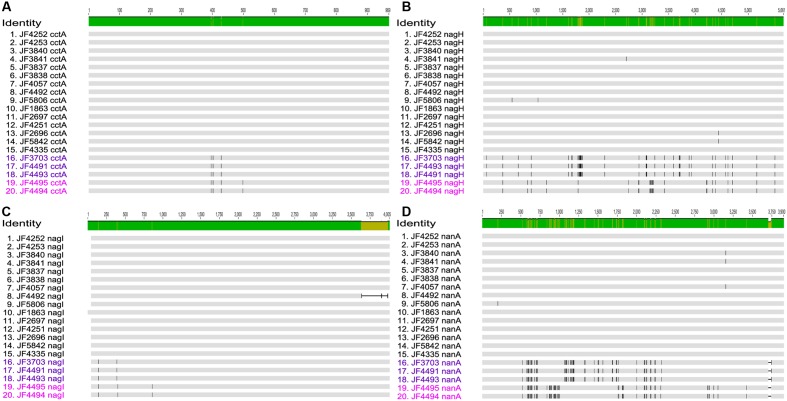
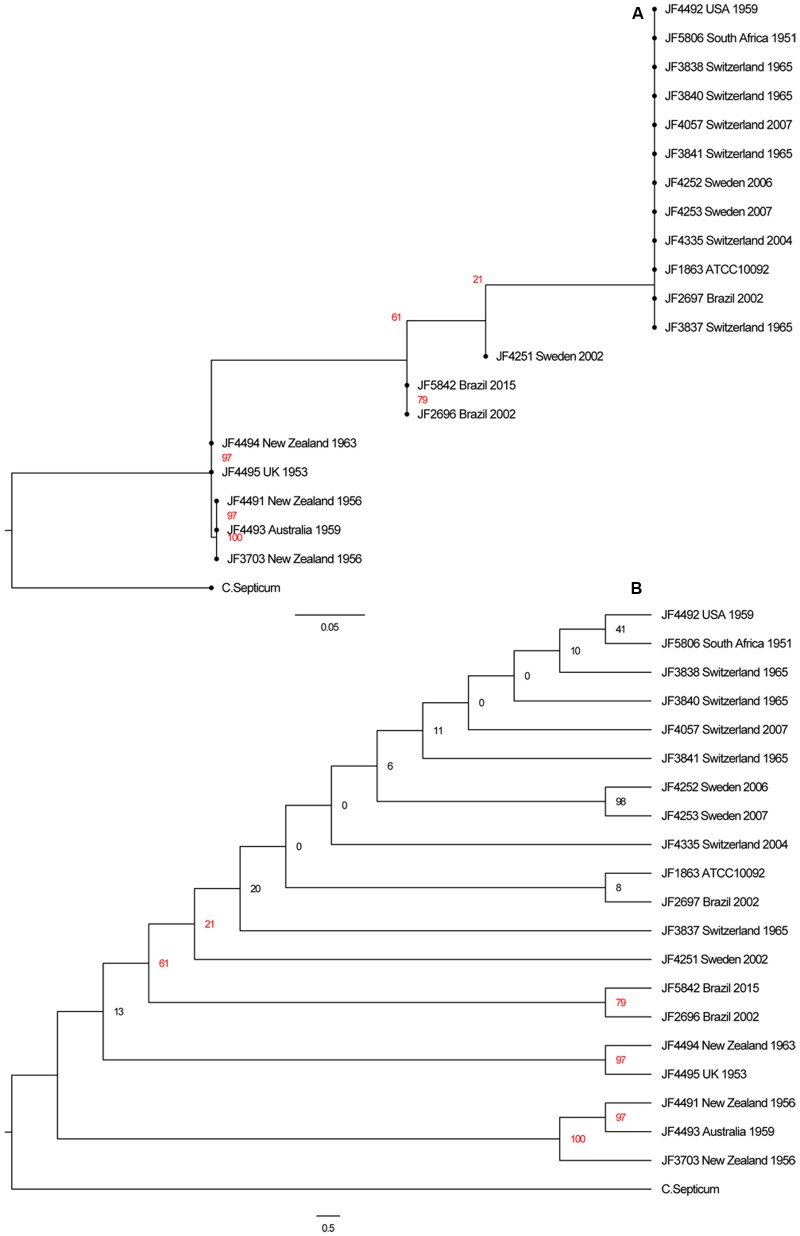
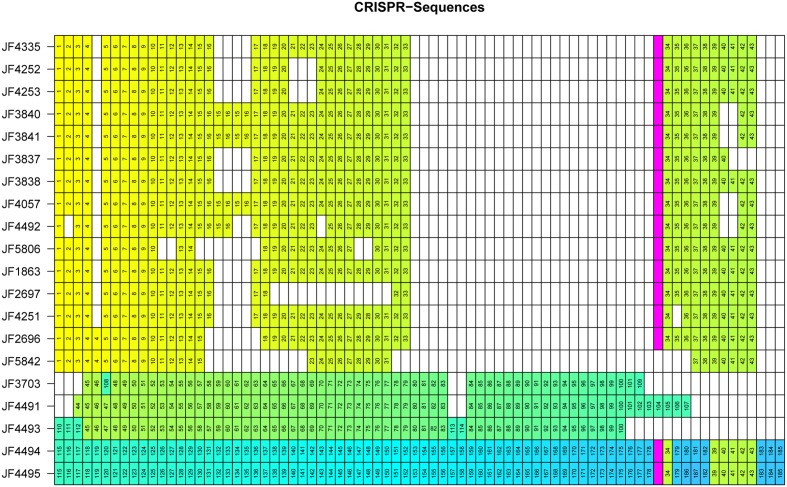
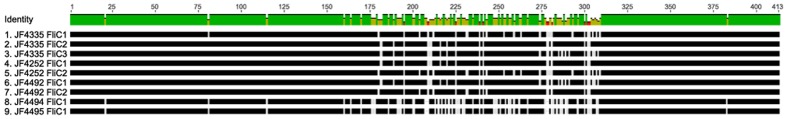
Full genome sequences of 20 strains of Clostridium chauvoei, the etiological agent of blackleg of cattle and sheep, isolated from four different continents over a period of 64 years (1951-2015) were determined and analyzed. The study reveals that the genome of the species C. chauvoei is highly homogeneous compared to the closely related species C. perfringens, a widespread pathogen that affects human and many animal species. Analysis of the CRISPR locus is sufficient to differentiate most C. chauvoei strains and is the most heterogenous region in the genome, containing in total 187 different spacer elements that are distributed as 30 - 77 copies in the various strains. Some genetic differences are found in the 3 allelic variants of fliC1, fliC2 and fliC3 genes that encode structural flagellin proteins, and certain strains do only contain one or two alleles. However, the major virulence genes including the highly toxic C.chauvoei toxin A, the sialidase and the two hyaluronidases are fully conserved as are the metabolic and structural genes of C. chauvoei. These data indicate that C. chauvoei is a strict ruminant-associated pathogen that has reached a dead end in its evolution.
Keywords: CRISPR; Clostridium chauvoei; blackleg; dead-end evolution; flagellin genes; virulence genes.
Figures
References
LinkOut - more resources
Full Text Sources
Other Literature Sources