

Primary Ciliary Dyskinesia: An Update on Clinical Aspects, Genetics, Diagnosis, and Future Treatment Strategies
- PMID: 28649564
- PMCID: PMC5465251
- DOI: 10.3389/fped.2017.00135
Primary Ciliary Dyskinesia: An Update on Clinical Aspects, Genetics, Diagnosis, and Future Treatment Strategies
Abstract
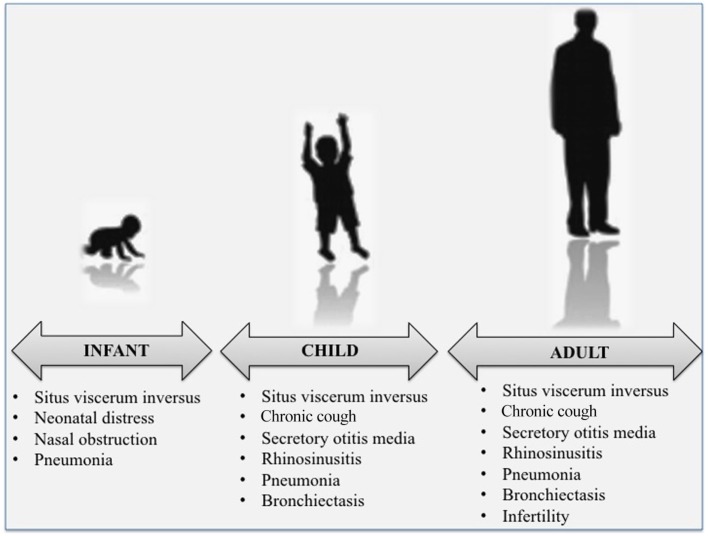
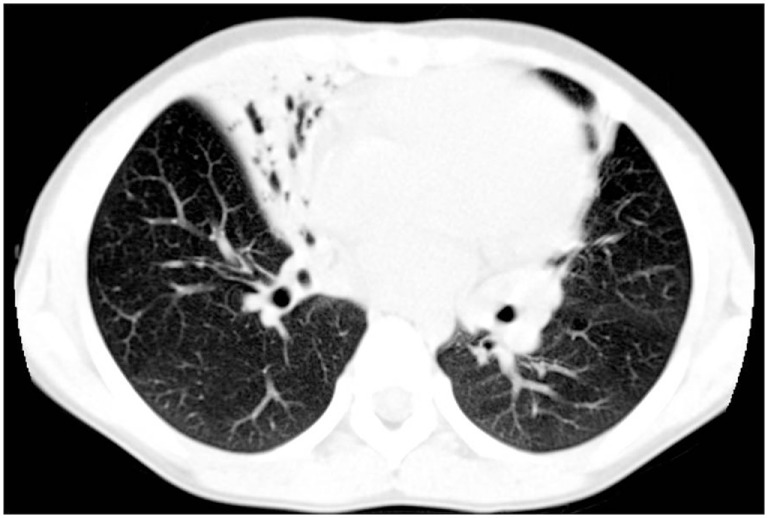
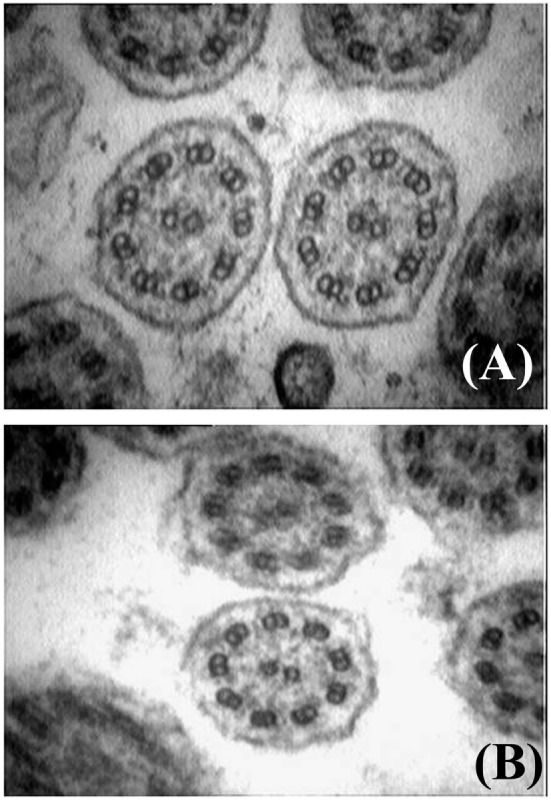
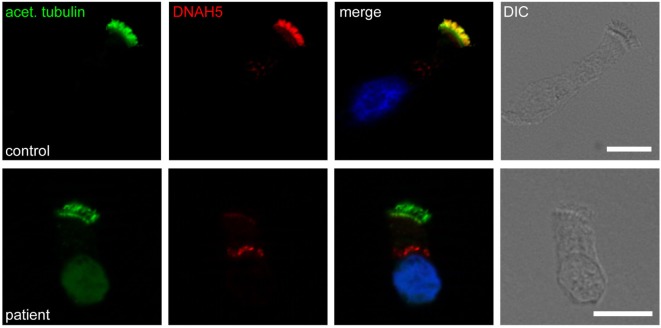
Primary ciliary dyskinesia (PCD) is an orphan disease (MIM 244400), autosomal recessive inherited, characterized by motile ciliary dysfunction. The estimated prevalence of PCD is 1:10,000 to 1:20,000 live-born children, but true prevalence could be even higher. PCD is characterized by chronic upper and lower respiratory tract disease, infertility/ectopic pregnancy, and situs anomalies, that occur in ≈50% of PCD patients (Kartagener syndrome), and these may be associated with congenital heart abnormalities. Most patients report a daily year-round wet cough or nose congestion starting in the first year of life. Daily wet cough, associated with recurrent infections exacerbations, results in the development of chronic suppurative lung disease, with localized-to-diffuse bronchiectasis. No diagnostic test is perfect for confirming PCD. Diagnosis can be challenging and relies on a combination of clinical data, nasal nitric oxide levels plus cilia ultrastructure and function analysis. Adjunctive tests include genetic analysis and repeated tests in ciliary culture specimens. There are currently 33 known genes associated with PCD and correlations between genotype and ultrastructural defects have been increasingly demonstrated. Comprehensive genetic testing may hopefully screen young infants before symptoms occur, thus improving survival. Recent surprising advances in PCD genetic designed a novel approach called "gene editing" to restore gene function and normalize ciliary motility, opening up new avenues for treating PCD. Currently, there are no data from randomized clinical trials to support any specific treatment, thus, management strategies are usually extrapolated from cystic fibrosis. The goal of treatment is to prevent exacerbations, slowing the progression of lung disease. The therapeutic mainstay includes airway clearance maneuvers mainly with nebulized hypertonic saline and chest physiotherapy, and prompt and aggressive administration of antibiotics. Standardized care at specialized centers using a multidisciplinary approach that imposes surveillance of lung function and of airway biofilm composition likely improves patients' outcome. Pediatricians, neonatologists, pulmonologists, and ENT surgeons should maintain high awareness of PCD and refer patients to the specialized center before sustained irreversible lung damage develops. The recent creation of a network of PCD clinical centers, focusing on improving diagnosis and treatment, will hopefully help to improve care and knowledge of PCD patients.
Keywords: Kartagener’s syndrome; bronchiectasis; ciliopathy; mucociliary clearance; primary ciliary dyskinesia.
Figures
References
-
- Kartagener M. Zur pathogenese der bronkiectasien. Bronkiectasien bei situs viscerum inversus. Beitr Klin Tuberk Spezif Tuberkuloseforsch (1933) 83:489–501. 10.1007/BF02141468 - DOI
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous