

An activating mutation of GNB1 is associated with resistance to tyrosine kinase inhibitors in ETV6-ABL1-positive leukemia
- PMID: 28650474
- PMCID: PMC5666322
- DOI: 10.1038/onc.2017.210
An activating mutation of GNB1 is associated with resistance to tyrosine kinase inhibitors in ETV6-ABL1-positive leukemia
Abstract
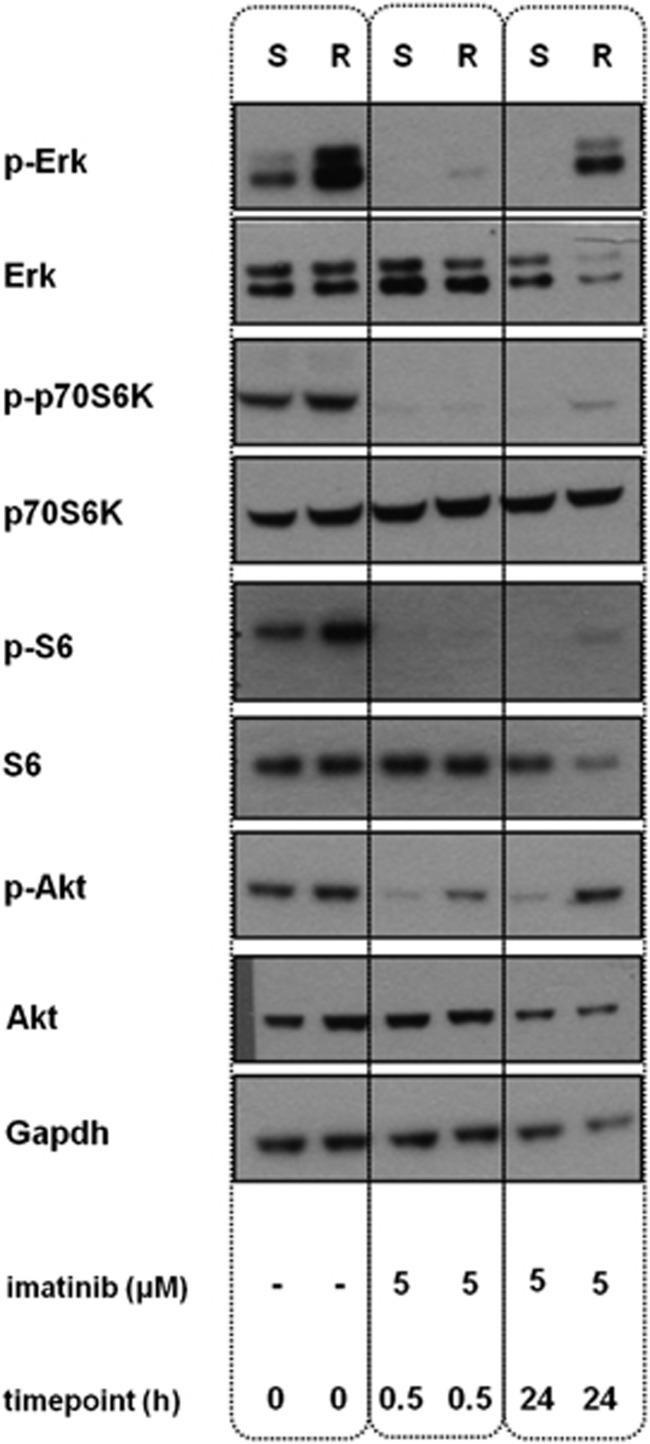
Leukemias harboring the ETV6-ABL1 fusion represent a rare subset of hematological malignancies with unfavorable outcomes. The constitutively active chimeric Etv6-Abl1 tyrosine kinase can be specifically inhibited by tyrosine kinase inhibitors (TKIs). Although TKIs represent an important therapeutic tool, so far, the mechanism underlying the potential TKI resistance in ETV6-ABL1-positive malignancies has not been studied in detail. To address this issue, we established a TKI-resistant ETV6-ABL1-positive leukemic cell line through long-term exposure to imatinib. ETV6-ABL1-dependent mechanisms (including fusion gene/protein mutation, amplification, enhanced expression or phosphorylation) and increased TKI efflux were excluded as potential causes of resistance. We showed that TKI effectively inhibited the Etv6-Abl1 kinase activity in resistant cells, and using short hairpin RNA (shRNA)-mediated silencing, we confirmed that the resistant cells became independent from the ETV6-ABL1 oncogene. Through analysis of the genomic and proteomic profiles of resistant cells, we identified an acquired mutation in the GNB1 gene, K89M, as the most likely cause of the resistance. We showed that cells harboring mutated GNB1 were capable of restoring signaling through the phosphoinositide-3-kinase (PI3K)/Akt/mTOR and mitogen-activated protein kinase (MAPK) pathways, whose activation is inhibited by TKI. This alternative GNB1K89M-mediated pro-survival signaling rendered ETV6-ABL1-positive leukemic cells resistant to TKI therapy. The mechanism of TKI resistance is independent of the targeted chimeric kinase and thus is potentially relevant not only to ETV6-ABL1-positive leukemias but also to a wider spectrum of malignancies treated by kinase inhibitors.
Conflict of interest statement
The authors declare no conflict of interest.
Figures






References
-
- Okuda K, Golub TR, Gilliland DG, Griffin JD. p210BCR/ABL, p190BCR/ABL, and TEL/ABL activate similar signal transduction pathways in hematopoietic cell lines. Oncogene 1996; 13: 1147–1152. - PubMed
-
- Hannemann JR, McManus DM, Kabarowski JH, Wiedemann LM. Haemopoietic transformation by the TEL/ABL oncogene. Br J Haematol 1998; 102: 475–485. - PubMed
-
- Voss J, Posern G, Hannemann JR, Wiedemann LM, Turhan AG, Poirel H et al. The leukaemic oncoproteins Bcr-Abl and Tel-Abl (ETV6/Abl) have altered substrate preferences and activate similar intracellular signalling pathways. Oncogene 2000; 19: 1684–1690. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
