

Unpaired Extracellular Cysteine Mutations of CSF3R Mediate Gain or Loss of Function
- PMID: 28652245
- PMCID: PMC5763920
- DOI: 10.1158/0008-5472.CAN-17-1052
Unpaired Extracellular Cysteine Mutations of CSF3R Mediate Gain or Loss of Function
Abstract
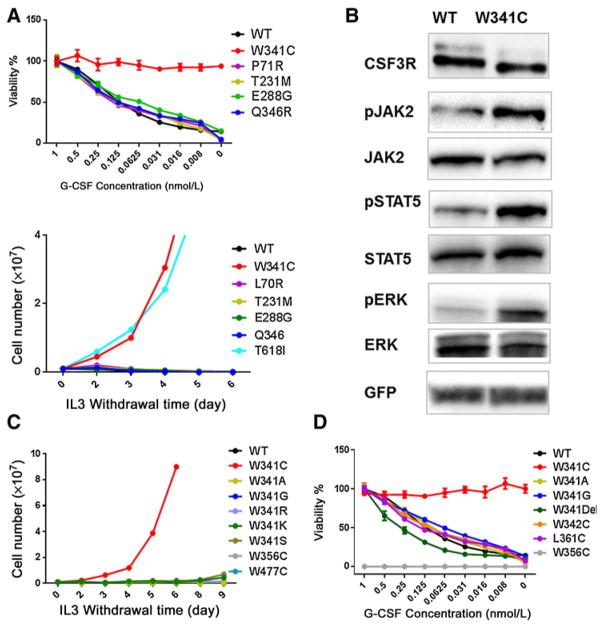
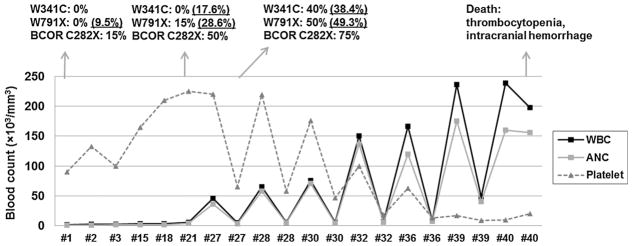
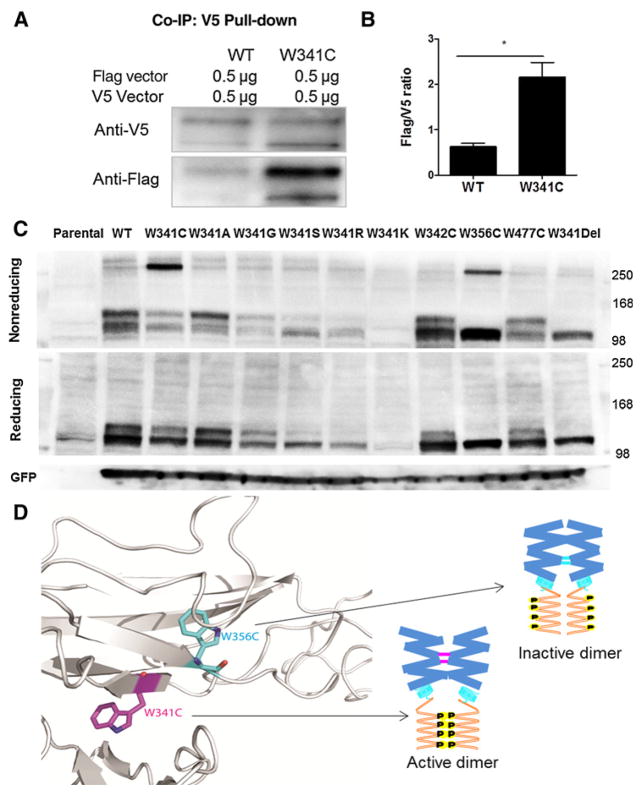
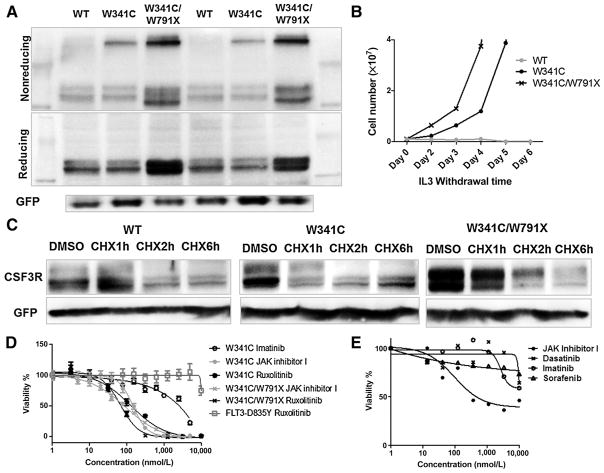
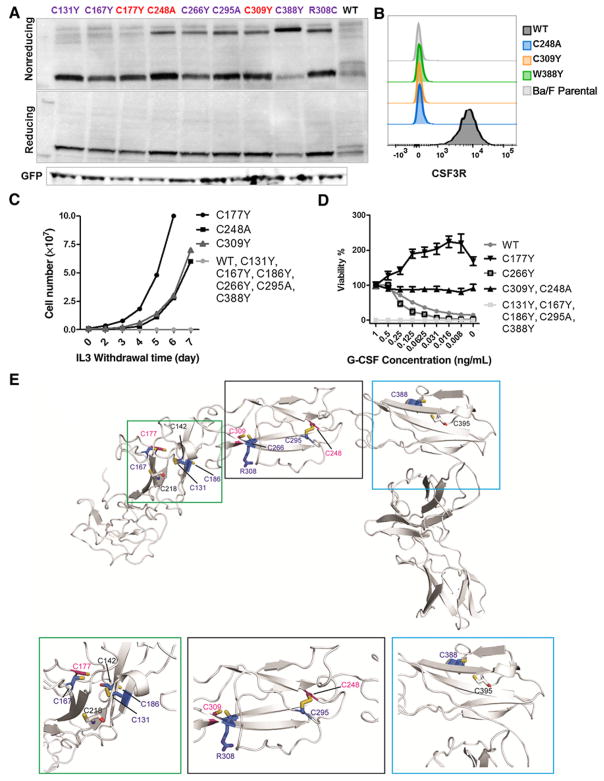
Exclusive of membrane-proximal mutations seen commonly in chronic neutrophilic leukemia (e.g., T618I), functionally defective mutations in the extracellular domain of the G-CSF receptor (CSF3R) have been reported only in severe congenital and idiopathic neutropenia patients. Here, we describe the first activating mutation in the fibronectin-like type III domain of the extracellular region of CSF3R (W341C) in a leukemia patient. This mutation transformed cells via cysteine-mediated intermolecular disulfide bonds, leading to receptor dimerization. Interestingly, a CSF3R cytoplasmic truncation mutation (W791X) found on the same allele as the extracellular mutation and the expansion of the compound mutation was associated with increased leukocytosis and disease progression of the patient. Notably, the primary patient sample and cells transformed by W341C and W341C/W791X exhibited sensitivity to JAK inhibitors. We further showed that disruption of original cysteine pairs in the CSF3R extracellular domain resulted in either gain- or loss-of-function changes, part of which was attributable to cysteine-mediated dimer formation. This, therefore, represents the first characterization of unpaired cysteines that mediate both gain- and loss-of-function phenotypes. Overall, our results show the structural and functional importance of conserved extracellular cysteine pairs in CSF3R and suggest the necessity for broader screening of CSF3R extracellular domain in leukemia patients. Cancer Res; 77(16); 4258-67. ©2017 AACR.
©2017 American Association for Cancer Research.
Conflict of interest statement
J.W. Tyner reports receiving commercial research grants from Agios, Aptose, Array, AstraZeneca, Constellation, Genentech, Incyte, Janssen, Seattle Genetics, Syros, and Takeda and is a consultant/advisory board member for Leap Oncology. No potential conflicts of interest were disclosed by the other authors.
Figures
References
-
- Aarts LHJ, Roovers O, Ward AC, Touw IP. Receptor activation and 2 distinct COOH-terminal motifs control G-CSF receptor distribution and internalization kinetics. Blood. 2004;103:571–9. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
